|
|||||


 OBETIDE OBETIDE
|
ELEA | |
| Semaglutida | ||
|
Hipoglucemiantes [Metabolismo] |
||
|
Venta Bajo Receta Solución Inyectable Industria Argentina |
||
|
||
|
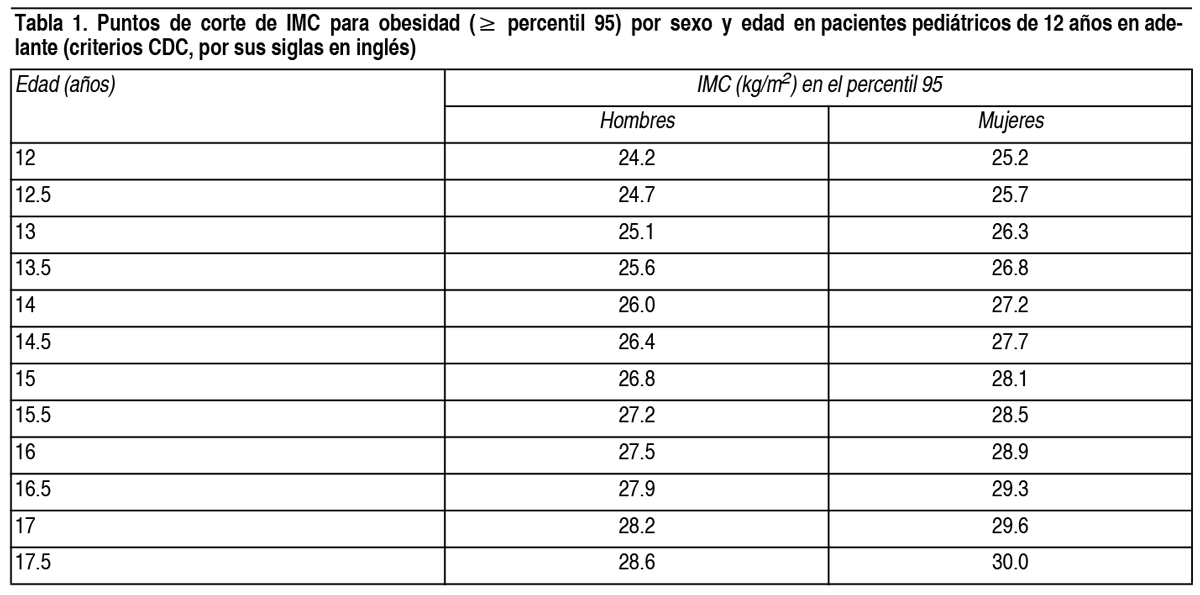
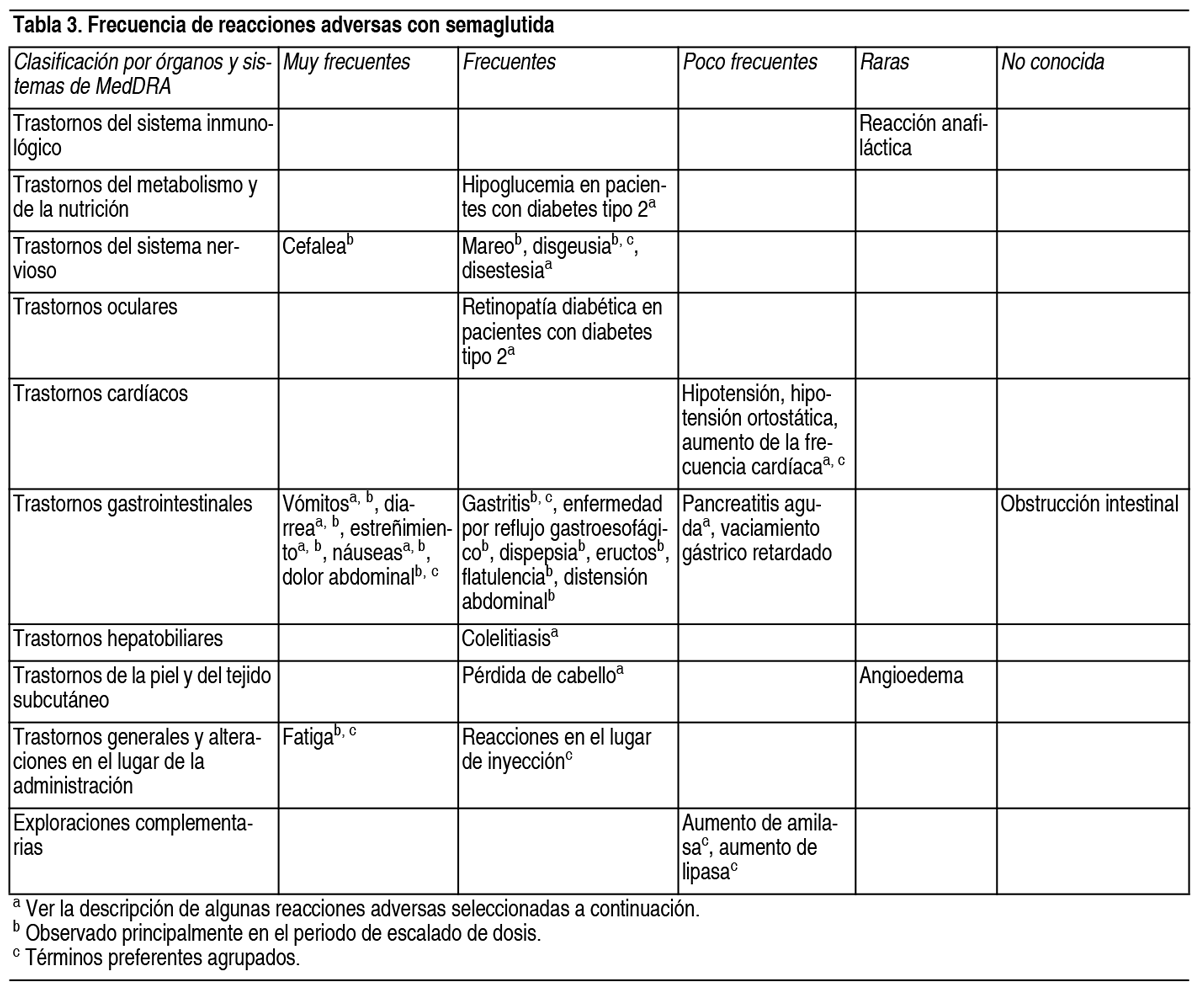
COMPOSICION: Semaglutida 0.25 mg/dosis-0.5 mg/dosis-1 mg/dosis: Cada ml de solución contiene: Principio activo: 1.34 mg de Semaglutida. Excipientes: Fosfato Disódico Dihidratado, Propilenglicol, Fenol, Ácido Clorhídrico/Hidróxido de Sodio c.s.p. pH 7-2-7.6, Agua para Inyectables. Semaglutida 1.7 mg/dosis-2.4 mg/dosis: Cada ml de solución contiene: Principio activo: 3.2 mg de Semaglutida. Excipientes: Fosfato Disódico Dihidratado, Propilenglicol, Fenol, Ácido Clorhídrico/Hidróxido de Sodio c.s.p. pH 7.2-7.6, Agua para Inyectables. ACCION: Fármacos utilizados en la diabetes, análogos del péptido similar al glucagón tipo 1 (GLP-1). Código ATC: A10BJ06. INDICACIONES: Adultos: Obetide está indicado como complemento a una dieta baja en calorías y a un aumento de la actividad física para el control de peso, incluida la pérdida y el mantenimiento del peso, en adultos con un índice de masa corporal (IMC) de: ≥30 kg/m2 (obesidad), o ≥27 kg/m2 a <30 kg/m2 (sobrepeso) en presencia de al menos 1 comorbilidad relacionada con el peso, p. ej., alteraciones de la glucemia (prediabetes o diabetes mellitus de tipo 2), hipertensión, dislipidemia, apnea obstructiva del sueño o enfermedad cardiovascular. Adolescentes (≥12 años): Obetide está indicado como complemento a una dieta baja en calorías y a un aumento de la actividad física para el control de peso en adolescentes de 12 años en adelante con obesidad* y peso corporal superior a 60 kg. Se debe discontinuar y reevaluar el tratamiento con Obetide si los pacientes adolescentes no han reducido al menos un 5 % su IMC tras 12 semanas en tratamiento con la dosis de 2.4 mg o la máxima dosis tolerada. *Obesidad (IMC≥ percentil 95) según se define en las tablas de crecimiento del IMC específicas por sexo y edad (CDC.gov) (ver Tabla 1). Ver Tabla DOSIS: Adultos: La dosis de mantenimiento de semaglutida 2.4 mg 1 vez a la semana se alcanza comenzando con una dosis de 0.25 mg. Para reducir la probabilidad de síntomas gastrointestinales, la dosis se debe escalar durante un período de 16 semanas hasta una dosis de mantenimiento de 2.4 mg 1 vez a la semana (ver Tabla 2). En caso de síntomas gastrointestinales significativos, se debe considerar retrasar el escalado de dosis o reducir a la dosis previa hasta que los síntomas hayan mejorado. No se recomiendan dosis semanales superiores a 2.4 mg. Tabla 2. Calendario de escalado de dosis: Escalado de dosis: Dosis semanal. Semana 1 a 4: 0.25 mg. Semana 5 a 8: 0.5 mg. Semana 9 a 12: 1 mg. Semana 13 a 16: 1.7 mg. Dosis de mantenimiento: 2.4 mg. Adolescentes: Se debe aplicar el mismo calendario de escalado de dosis para adolescentes de 12 años en adelante (ver Tabla 2). La dosis se debe de incrementar hasta 2.4 mg (dosis de mantenimiento) o hasta que se alcance la máxima dosis tolerada. No se recomiendan dosis semanales superiores a 2.4 mg. Pacientes con diabetes tipo 2: Al iniciar semaglutida en pacientes con diabetes tipo 2, se debe considerar reducir la dosis de insulina o de secretagogos de insulina (como sulfonilureas) que se administran de forma concomitante para reducir el riesgo de hipoglucemia. Dosis olvidadas: Si se olvida una dosis, esta se debe administrar tan pronto como sea posible y dentro de los 5 días posteriores a la dosis olvidada. En caso de que hayan transcurrido más de 5 días, se debe saltar la dosis olvidada y la siguiente dosis se debe administrar de forma habitual en el día programado. En cualquiera de los casos, los pacientes pueden reanudar a continuación su esquema de dosificación habitual de 1 vez a la semana. Si se olvidan más dosis, se debe considerar reducir a la dosis de inicio para volver a comenzar. Poblaciones especiales: Pacientes de edad avanzada (≥65 años): No se requiere un ajuste de la dosis en función de la edad. La experiencia clínica en pacientes de ≥ 85 años es limitada. Pacientes con insuficiencia renal: No se requiere un ajuste de dosis en pacientes con insuficiencia renal leve o moderada. La experiencia con el uso de semaglutida en pacientes con insuficiencia renal grave es limitada. No se recomienda el uso de semaglutida en pacientes con insuficiencia renal grave (TFGe <30 ml/min/1.73 m2), incluidos pacientes con enfermedad renal terminal. Pacientes con insuficiencia hepática: No se requiere un ajuste de dosis en pacientes con insuficiencia hepática leve o moderada. La experiencia con el uso de semaglutida en pacientes con insuficiencia hepática grave es limitada. No se recomienda el uso de semaglutida en pacientes con insuficiencia hepática grave y se debe usar con precaución en pacientes con insuficiencia hepática leve o moderada. Población pediátrica: No se requiere ajuste de dosis en adolescentes de 12 años en adelante. No se ha establecido todavía la seguridad y eficacia de semaglutida en niños menores de 12 años. Forma de administración: Vía subcutánea. Obetide se administra 1 vez a la semana a cualquier hora del día, con o sin alimentos. Se debe inyectar por vía subcutánea en el abdomen, el muslo o la parte superior del brazo. El lugar de la inyección se puede cambiar. No se debe administrar por vía intravenosa ni intramuscular. El día de administración semanal se puede cambiar si es necesario, siempre que el tiempo entre 2 dosis sucesivas sea de al menos 3 días (>72 horas). Una vez seleccionado el nuevo día de administración, se debe proseguir con la pauta de dosificación de 1 vez a la semana. Al administrar Obetide, jeringa prellenada de 1 solo uso, la jeringa se debe presionar firmemente contra la piel hasta que el émbolo se detenga. Se debe advertir a los pacientes que deben leer detenidamente las instrucciones de uso incluidas en el prospecto antes de administrar el medicamento. CONTRAINDICACIONES: Hipersensibilidad al principio activo o a cualquiera de los componentes de la formulación. ADVERTENCIAS: Aspiración en asociación con anestesia general o sedación profunda: Se han notificado casos de aspiración pulmonar en pacientes que recibieron agonistas de receptores GLP-1 sometidos a anestesia general o sedación profunda. Por consiguiente, se debe considerar el aumento del riesgo de contenido gástrico residual debido al retraso en el vaciado gástrico antes de realizar los procedimientos con anestesia general o sedación profunda. Deshidratación: El uso de los agonistas del receptor de GLP-1 puede estar asociado a reacciones adversas gastrointestinales que pueden causar deshidratación, que en algunos casos raros pueden dar lugar a un deterioro de la función renal. Se debe advertir a los pacientes de que existe un riesgo potencial de deshidratación relacionado con los efectos adversos gastrointestinales y de que tomen precauciones para evitar la pérdida de líquidos. Pancreatitis aguda: Se ha observado pancreatitis aguda con el uso de agonistas del receptor de GLP-1. Se debe informar a los pacientes de los síntomas característicos de la pancreatitis aguda. Ante la sospecha de pancreatitis, se debe interrumpir el tratamiento con semaglutida y no se debe reanudar si se confirma la pancreatitis. Se debe extremar la precaución en pacientes con antecedentes de pancreatitis. En ausencia de otros signos y síntomas de la pancreatitis aguda, las elevaciones de las enzimas pancreáticas por sí solas no son un factor predictivo de la pancreatitis aguda. Pacientes con diabetes tipo 2: No se debe utilizar semaglutida como sustituto de la insulina en pacientes con diabetes tipo 2. No se debe utilizar semaglutida en combinación con otros productos agonistas del receptor de GLP-1. No se ha evaluado y se considera probable un mayor riesgo de reacciones adversas relacionadas con la sobredosis. Hipoglucemia en pacientes con diabetes tipo 2: Se sabe que la insulina y sulfonilurea provocan hipoglucemia. Los pacientes tratados con semaglutida en combinación con una sulfonilurea o insulina podrían presentar un riesgo mayor de hipoglucemia. Es posible disminuir el riesgo de hipoglucemia reduciendo la dosis de sulfonilurea o de insulina al inicio del tratamiento con un agonista del receptor de GLP-1. Retinopatía diabética en pacientes con diabetes tipo 2: En pacientes con retinopatía diabética tratados con semaglutida, se ha observado un mayor riesgo de desarrollar complicaciones de la retinopatía diabética. La rápida mejora del control glucémico se ha asociado con un empeoramiento temporal de la retinopatía diabética, pero no se pueden excluir otros mecanismos. Se debe controlar estrechamente a los pacientes con retinopatía diabética que utilizan semaglutida y tratarlos según las directrices clínicas correspondientes. No hay experiencia con semaglutida en pacientes con diabetes tipo 2 con retinopatía diabética no controlada o potencialmente inestable. No se recomienda el tratamiento con Obetide en estos pacientes. Poblaciones no estudiadas: No se ha investigado la seguridad y la eficacia de semaglutida en pacientes: Tratados con otros productos para el control de peso. Con diabetes tipo 1. Con insuficiencia renal grave. Con insuficiencia hepática grave. Con insuficiencia cardíaca congestiva de clase IV según la Asociación del Corazón de Nueva York (NYHA, por sus siglas en inglés). No se recomienda el uso en estos pacientes. Existe una experiencia limitada con semaglutida en pacientes: De 85 años o más. Con insuficiencia hepática leve o moderada. Con enfermedad inflamatoria intestinal. Con gastroparesia diabética. Usar con precaución en estos pacientes. EFECTOS: Resumen del perfil de seguridad: En 4 ensayos clínicos, en fase 3a, se expusieron 2650 pacientes adultos a semaglutida. La duración de los ensayos fue de 68 semanas. Las reacciones adversas comunicadas con más frecuencia fueron los trastornos gastrointestinales, como náuseas, diarrea, estreñimiento y vómitos. Tabla de reacciones adversas: En la Tabla 3 se enumeran las reacciones adversas identificadas en los ensayos clínicos en adultos y en etapas de poscomercialización. Las frecuencias están basadas en un conjunto de ensayos en fase 3a. A continuación, se indican las reacciones adversas asociadas a semaglutida según la clasificación por órganos y sistemas y la frecuencia. Las categorías de frecuencias se definen del siguiente modo: Muy frecuentes (≥1/10), frecuentes (≥1/100 a <1/10), poco frecuentes (≥1/1000 a <1/100), raras (≥1/10 000 a <1/1000), muy raras (<1/10 000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Ver Tabla Reacciones adversas gastrointestinales: Durante el periodo del ensayo de 68 semanas, se produjeron náuseas en el 43.9 % de los pacientes tratados con semaglutida (16.1 % para placebo), diarrea en el 29.7 % (15.9 % para placebo) y vómitos en el 24.5 % (6.3 % para placebo). La mayoría de los acontecimientos fueron de leves a moderados en gravedad y de corta duración. Se produjo estreñimiento en el 24.2 % de los pacientes tratados con semaglutida (11.1 % para placebo) y fue de gravedad leve a moderada y de mayor duración. En los pacientes tratados con semaglutida, la mediana de duración de las náuseas fue de 8 días, de los vómitos 2 días, de la diarrea 3 días y del estreñimiento 47 días. Los pacientes con insuficiencia renal moderada (TFGe ≥30 a <60 ml/min/1.73 m2) pueden presentar más efectos gastrointestinales cuando reciben tratamiento con semaglutida. Los acontecimientos gastrointestinales condujeron a la interrupción permanente del tratamiento en el 4.3 % de los pacientes. Pancreatitis aguda: La frecuencia de pancreatitis aguda confirmada por adjudicación notificada en ensayos clínicos en fase 3a fue del 0.2 % para semaglutida y <0.1 % para placebo, respectivamente. En estudios clínicos, ensayo de resultados cardiovasculares, la frecuencia de pancreatitis aguda confirmada por adjudicación fue del 0.2 % para semaglutida y del 0.3 % para el placebo. Enfermedad aguda por cálculos biliares/colelitiasis: Se notificó colelitiasis en el 1.6 % y provocó colecistitis en el 0.6 % de los pacientes tratados con semaglutida. Se notificaron colelitiasis y colecistitis en el 1.1 % y el 0.3 %, respectivamente, de los pacientes tratados con placebo. Pérdida de cabello: Se notificó pérdida de cabello en el 2.5 % de los pacientes tratados con semaglutida y en el 1.0 % de los pacientes tratados con placebo. Los acontecimientos fueron principalmente de intensidad leve y la mayoría de los pacientes se recuperaron mientras seguían en tratamiento. La pérdida de cabello se notificó con más frecuencia en pacientes con una mayor pérdida de peso (≥20 %). Aumento de la frecuencia cardíaca: En los ensayos en fase 3a, se observó un aumento medio de 3 latidos por minuto (lpm) desde una media al inicio de 72 lpm en los pacientes tratados con semaglutida. Las proporciones de sujetos con un aumento del pulso desde el valor inicial ≥10 lpm en cualquier punto durante el periodo de tratamiento fueron del 67 % en el grupo de semaglutida frente al 50.1 % en el grupo de placebo. Inmunogenicidad: En línea con las propiedades potencialmente inmunogénicas de los medicamentos que contienen proteínas o péptidos, en un ensayo clínico se observó la posibilidad de que los pacientes desarrollaran anticuerpos durante el tratamiento con semaglutida. La proporción de pacientes con un resultado positivo en el análisis de anticuerpos antisemaglutida en cualquier punto temporal posterior al inicio del estudio fue baja (2.9 %). Al final del ensayo, no se detectaron pacientes con anticuerpos neutralizantes antisemaglutida o anticuerpos antisemaglutida con efecto neutralizante del GLP-1 endógeno. Se señala que, durante el tratamiento, las elevadas concentraciones de semaglutida podrían haber limitado la sensibilidad de los ensayos, por lo que no se puede descartar completamente la existencia de resultados falsos negativos. No obstante, la presencia de anticuerpos fue transitoria y no se evidenció una repercusión aparente en la eficacia y la seguridad en aquellos sujetos que presentaron resultados positivos a anticuerpos durante y después del tratamiento. Hipoglucemia en pacientes con diabetes tipo 2: En un estudio clínico, se observó hipoglucemia clínicamente significativa en el 6.2 % (0.1 acontecimientos/paciente año) de los sujetos tratados con semaglutida en comparación con el 2.5 % (0.03 acontecimientos/paciente año) de los sujetos tratados con placebo. La hipoglucemia con semaglutida se observó tanto con el uso simultáneo de sulfonilurea como sin él. Un episodio (0.2 % de los pacientes, 0.002 acontecimientos/paciente año) se notificó como grave en un sujeto no tratado de forma simultánea con una sulfonilurea. El riesgo de hipoglucemia se incrementó cuando semaglutida se usó con una sulfonilurea. En otro estudio clínico de referencia, se observó hipoglucemia clínicamente significativa en el 4.2 % de los sujetos, tanto en el grupo de semaglutida como en el placebo, cuando se utilizó en combinación con sulfonilurea y/o insulina (0.065 acontecimientos/paciente año con semaglutida y 0.098 acontecimientos/paciente año con placebo). Retinopatía diabética en pacientes con diabetes tipo 2: En un ensayo clínico de 2 años, se investigó semaglutida 0.5 mg y 1 mg frente a placebo en 3297 pacientes con diabetes tipo 2, alto riesgo cardiovascular, diabetes de larga duración y mal control de la glucemia. En este ensayo, se produjeron acontecimientos adjudicados de complicaciones de la retinopatía diabética en más pacientes tratados con semaglutida (3.0 %) en comparación con placebo (1.8 %). Esto se observó en pacientes con retinopatía diabética conocida tratados con insulina. La diferencia de tratamiento apareció pronto y persistió a lo largo del ensayo. En otro estudio clínico, se notificaron trastornos en la retina en el 6.9 % de pacientes tratados con semaglutida, el 6.2 % de los pacientes tratados con semaglutida 1 mg y el 4.2 % de los pacientes tratados con placebo. La mayoría de estos acontecimientos se notificaron como retinopatía diabética (4.0 %, 2.7 % y 2.7 %, respectivamente) y retinopatía no proliferativa (0.7 %, 0 % y 0 %, respectivamente). Disestesia: Se notificaron acontecimientos relacionados con un cuadro clínico de la sensación cutánea alterada como parestesia, dolor en la piel, piel sensible, disestesia y sensación de ardor en la piel en el 2.1 % de pacientes tratados con semaglutida 2.4 mg, y en un 1.2 % de pacientes tratados con placebo. Los acontecimientos fueron de leves a moderados en gravedad y la mayoría de los pacientes se recuperaron mientras continuaban con el tratamiento. Población pediátrica: En un ensayo clínico se expuso a semaglutida a 133 pacientes adolescentes de 12 a 18 años, con obesidad o con sobrepeso, con al menos 1 comorbilidad relacionada con el peso. La duración del ensayo fue de 68 semanas. En general, la frecuencia, el tipo y la gravedad de las reacciones adversas en adolescentes fueron comparables a las observadas en la población adulta. Se notificó colelitiasis en un 3.8 % de los pacientes tratados con semaglutida y en un 0 % de los pacientes tratados con placebo. No se han encontrado efectos en el crecimiento o en el desarrollo puberal tras 68 semanas de tratamiento. Otras poblaciones especiales: En ensayos clínicos de referencia, el perfil de reacciones adversas en adultos con enfermedad cardiovascular establecida fue similar al observado en los ensayos de control de peso de fase 3a. En ensayos clínicos en adultos con insuficiencia cardíaca relacionada con obesidad, con fracción de eyección preservada (HFpEF, por sus siglas en inglés), el perfil de las reacciones adversas fue similar al observado en los ensayos de fase 3a sobre el control de peso. Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales de la salud a notificar las sospechas de reacciones adversas al Departamento de Farmacovigilancia del Laboratorio Elea enviando un correo electrónico a farmacovigilancia@elea.com o telefónicamente al 0800 333 3532. PRECAUCIONES: Fertilidad, embarazo y lactancia: Mujeres en edad fértil: Se recomienda que las mujeres en edad fértil utilicen métodos anticonceptivos durante el tratamiento con semaglutida. Embarazo: Los ensayos realizados en animales han mostrado toxicidad reproductiva. Los datos relativos al uso de semaglutida en mujeres embarazadas son limitados. Por tanto, no se debe utilizar semaglutida durante el embarazo. Se debe interrumpir el tratamiento con semaglutida en caso de que una paciente desee quedarse embarazada o si se produce un embarazo. Debido a la larga semivida de semaglutida, el tratamiento se debe interrumpir al menos 2 meses antes de un embarazo planeado. Lactancia: En ratas lactantes, semaglutida se excretó en la leche materna. No se puede excluir el riesgo en lactantes. Semaglutida no se debe utilizar durante la lactancia. Fertilidad: Se desconoce el efecto de semaglutida sobre la fertilidad en los seres humanos. En el caso de las ratas, semaglutida no afectó a la fertilidad de los machos. En el caso de las ratas hembra, se observó un aumento de la duración del ciclo estral y una ligera disminución del número de ovulaciones en dosis asociadas con la pérdida de peso corporal materno. Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de semaglutida sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Sin embargo, se pueden experimentar mareos principalmente durante el periodo de escalado de dosis. En el caso de que se produzcan mareos, la conducción o el uso de máquinas se deben realizar con precaución. Pacientes con diabetes tipo 2: Si semaglutida se utiliza en combinación con una sulfonilurea o insulina, se debe advertir a los pacientes que extremen las precauciones para evitar una hipoglucemia mientras conducen y utilizan máquinas. INTERACCIONES: Semaglutida retrasa el vaciamiento gástrico y podría influir potencialmente en la absorción de medicamentos administrados de forma concomitante por vía oral. No se observó ningún efecto clínicamente relevante sobre la velocidad de vaciamiento gástrico con semaglutida 2.4 mg, probablemente debido a un efecto de tolerancia. Semaglutida se debe utilizar con precaución en pacientes en tratamiento con medicamentos orales que requieren una absorción gastrointestinal rápida. Paracetamol: Semaglutida retrasa la velocidad de vaciamiento gástrico tal como determinó la farmacocinética de paracetamol durante una prueba de comida estándar. El AUC0-60 min y la Cmáx de paracetamol se redujeron en un 27 % y un 23 %, respectivamente, tras el uso concomitante de semaglutida 1 mg. La exposición total de paracetamol (AUC0-5 h) no se vio afectada. No se observó ningún efecto clínicamente relevante en el paracetamol con semaglutida. No es necesario un ajuste de dosis de paracetamol cuando se administra con semaglutida. Anticonceptivos orales: No se prevé que semaglutida disminuya la eficacia de los anticonceptivos orales. Tras la administración concomitante de un medicamento anticonceptivo oral combinado (0.03 mg de etinilestradiol/0.15 mg de levonorgestrel) y semaglutida, no se modificó de una forma clínicamente significativa la exposición general de etinilestradiol ni de levonorgestrel. La exposición de etinilestradiol no se vio afectada; se observó un aumento del 20 % en la exposición de levonorgestrel en estado estacionario. La Cmáx no se vio afectada por ninguno de los compuestos. Atorvastatina: Semaglutida no modificó la exposición general de atorvastatina tras la administración de una dosis única de atorvastatina (40 mg). La Cmáx de atorvastatina se redujo en un 38 %. Se concluyó que esto no era clínicamente significativo. Digoxina: Semaglutida no modificó la exposición general ni la Cmáx de digoxina tras la administración de una dosis única de digoxina (0.5 mg). Metformina: Semaglutida no modificó la exposición general ni la Cmáx de metformina tras la administración de 500 mg de metformina 2 veces al día durante 3.5 días. Warfarina y otros derivados de la cumarina: Semaglutida no modificó la exposición general ni la Cmáx de los enantiómeros R y S de warfarina tras la administración de una dosis única de warfarina (25 mg) y los efectos farmacodinámicos de warfarina, determinados por índice internacional normalizado (INR, por sus siglas en inglés), no se vieron afectados de una forma clínicamente significativa. Sin embargo, se han notificado casos de INR disminuido durante el uso concomitante con acenocumarol y semaglutida. Se recomienda el control frecuente del INR al iniciar el tratamiento con semaglutida en pacientes con warfarina u otros derivados de la cumarina. Población pediátrica: Solo se han realizado estudios de interacción en adultos. SOBREDOSIFICACION: Una sobredosis con semaglutida se puede asociar a trastornos gastrointestinales que pueden causar deshidratación. En caso de sobredosis, se debe observar al paciente por si presentara signos clínicos y se debe iniciar el tratamiento de soporte adecuado. Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los centros de toxicología: Hospital de Pediatría «Ricardo Gutiérrez»: (011) 4962 6666 / 2247. Hospital «A. Posadas»: (011) 4654 6648 / 4658 7777. Centro Nacional de Intoxicaciones, Tel.: 0800 333 0160. Para otras consultas: Centro de Atención Telefónica de Laboratorio Elea: 0800 333 3532. PROPIEDADES: Mecanismo de acción: Semaglutida es un análogo de GLP-1 con un 94 % de homología de secuencia con el GLP-1 humano. Semaglutida actúa como un agonista del receptor de GLP-1 que se une de forma selectiva al receptor de GLP-1, el objetivo del GLP-1 nativo, y lo activa. GLP-1 es un regulador fisiológico de apetito y de la ingesta de calorías. Está presente en varias áreas del cerebro implicadas en la regulación del apetito. Estudios en animales muestran que semaglutida actúa en el cerebro a través del receptor de GLP-1. Semaglutida tiene efectos directos en áreas del cerebro implicadas en la regulación homeostática de la ingesta de alimentos en el hipotálamo y el tronco encefálico. Semaglutida puede afectar al sistema hedónico de recompensa a través de efectos directos e indirectos en áreas del cerebro, incluido el septo, el tálamo y la amígdala. Ensayos clínicos muestran que semaglutida reduce la ingesta calórica, aumenta la sensación de saciedad, plenitud y el control de la ingesta, reduce la sensación de hambre, y la frecuencia y la intensidad del ansia de comer. Además, semaglutida reduce la preferencia por alimentos con alto contenido en grasa. Semaglutida organiza las contribuciones homeostáticas y hedónicas con función ejecutiva para regular la ingesta calórica, el apetito, la recompensa y la elección de alimentos. Además, semaglutida ha demostrado en ensayos clínicos que reduce la glucosa en sangre de un modo dependiente de la glucosa, mediante la estimulación de la secreción de insulina y reduciendo la secreción de glucagón cuando la glucemia es elevada. El mecanismo de disminución de la glucemia también implica un ligero retraso en el vaciamiento gástrico en la fase posprandial temprana. Durante la hipoglucemia, semaglutida disminuye la secreción de insulina y no afecta a la secreción de glucagón. Los receptores de GLP-1 también se expresan en el corazón, la vasculatura, el sistema inmunitario y los riñones. En ensayos clínicos, semaglutida tiene un efecto beneficioso en lípidos plasmáticos, disminuye la presión arterial sistólica y reduce la inflamación. Asimismo, ensayos en animales han demostrado que semaglutida atenuó el desarrollo de aterosclerosis y tuvo una acción antiinflamatoria en el sistema cardiovascular. El mecanismo de acción de semaglutida para la reducción del riesgo cardiovascular es probablemente multifactorial, en parte impulsado por los efectos de pérdida de peso y los efectos sobre factores de riesgo cardiovascular conocidos (reducción de la presión arterial, mejora en el perfil lipídico y metabolismo de la glucosa, y efectos antiinflamatorios, demostrados por las reducciones en el nivel de proteína C reactiva de alta sensibilidad [hsCRP por sus siglas en inglés]). No se ha establecido el mecanismo exacto para la reducción del riesgo cardiovascular. Propiedades farmacodinámicas: Apetito, ingesta calórica y elecciones alimentarias: Semaglutida reduce el apetito al aumentar la sensación de plenitud y de saciedad, mientras se reduce el hambre y el posible consumo de alimentos. En un ensayo de fase 1, la ingesta calórica durante una comida ad libitum fue un 35 % menor con semaglutida en comparación con placebo después de 20 semanas de la administración de la dosis. Esto fue respaldado por un mejor control de la alimentación, menos ansia de comer y una preferencia relativamente menor por los alimentos con alto contenido en grasa. En un estudio clínico, se evaluó de manera adicional el ansia de comer con un Cuestionario de Control de la Alimentación (CoEQ, por sus siglas en inglés). En la semana 104, la diferencia del tratamiento estimado tanto para el control del ansia de comer y de las ansias de comer comida salada favoreció significativamente a semaglutida, mientras que no se observó un efecto claro en las ansias de comer comida dulce. Lípidos en ayunas y posprandiales: Semaglutida 1 mg, en comparación con el placebo, redujo las concentraciones de triglicéridos y lipoproteínas de muy baja densidad (VLDL, por sus siglas en inglés) en un 12 % y un 21 %, respectivamente. La respuesta posprandial de los triglicéridos y las VLDL a una comida rica en grasas se redujo en >40 %. Propiedades farmacocinéticas: En comparación con el GLP-1 nativo, semaglutida tiene una semivida prolongada de aproximadamente 1 semana, por lo que es adecuada para la administración subcutánea 1 vez a la semana. El mecanismo principal de protracción es la unión a albúmina, que propicia una disminución del aclaramiento renal y protege de la degradación metabólica. Asimismo, semaglutida es resistente frente a la degradación por la enzima DPP-4. Absorción: La concentración media de semaglutida en estado estacionario tras la administración s.c. de la dosis de mantenimiento de semaglutida fue de aproximadamente 75 nmol/l en pacientes con sobrepeso (IMC ≥27 kg/m2 a <30 kg/m2) u obesidad (IMC ≥30 kg/m2) según los datos de los ensayos en fase 3a, donde el 90 % de los pacientes tenían una concentración media entre 51 nmol/l y 110 nmol/l. La exposición en estado estacionario de semaglutida aumentó proporcionalmente con dosis de 0.25 mg a 2.4 mg 1 vez a la semana. La exposición en estado estacionario fue estable con el tiempo según lo evaluado hasta la semana 68. Asimismo, se logró una exposición similar con la administración s.c. de semaglutida en el abdomen, el muslo o la parte superior de brazo. La biodisponibilidad absoluta de semaglutida fue del 89 %. Distribución: El volumen de distribución medio de semaglutida tras la administración intravenosa en pacientes con sobrepeso u obesidad fue de aproximadamente 12.4 litros. Semaglutida se encuentra ampliamente unida a albumina en el plasma (>99 %). Metabolismo/Biotransformación: Antes de la excreción, semaglutida se metaboliza en gran medida mediante proteólisis del esqueleto peptídico y beta-oxidación secuencial de la cadena lateral del ácido graso. La enzima endopeptidasa neutra (NEP, por sus siglas en inglés) fue identificada como una de las enzimas metabólicas activas. Eliminación: Las principales vías de la excreción de material relacionado con semaglutida son a través de la orina y las heces. Alrededor del 3 % de la dosis absorbida se excretó en forma de semaglutida intacta en la orina. El aclaramiento de semaglutida en pacientes con sobrepeso (IMC ≥27 kg/m2 a <30 kg/m2) u obesidad (IMC ≥30 kg/m2) fue de aproximadamente 0.05 l/h. Con una semivida de eliminación aproximada de 1 semana, semaglutida permanecerá en la circulación durante un tiempo aproximado de 7 semanas después de la última dosis de 2.4 mg. Poblaciones especiales: Pacientes de edad avanzada: La edad no tuvo ningún efecto sobre las propiedades farmacocinéticas de semaglutida en pacientes de 20-86 años. Sexo y origen étnico: El sexo y el origen étnico no tuvieron ningún efecto sobre las propiedades farmacocinéticas de semaglutida. Peso corporal: El peso corporal tuvo un efecto en la exposición de semaglutida. Un mayor peso corporal se asoció a una menor exposición; una diferencia del 20 % en el peso corporal de las personas se traducirá en una diferencia aproximada del 18 % en la exposición. La dosis semanal de 2.4 mg de semaglutida proporcionó exposiciones sistémicas adecuadas en el rango de peso corporal de 54.4 a 245.6 kg evaluado para la respuesta a la exposición en los ensayos clínicos. Insuficiencia renal: La insuficiencia renal no tuvo ningún efecto clínicamente significativo sobre la farmacocinética de semaglutida. Esto se constató comparando los efectos de una dosis única de 0.5 mg de semaglutida en pacientes con diversos grados de insuficiencia renal (leve, moderada, grave o pacientes en diálisis) en comparación con pacientes con función renal normal. Esto también se mostró para pacientes con sobrepeso (IMC ≥27 kg/m2 a <30 kg/m2) u obesidad (IMC ≥30 kg/m2) e insuficiencia renal de leve a moderada. Insuficiencia hepática: La insuficiencia hepática no tuvo ningún efecto en la exposición de semaglutida. La farmacocinética de semaglutida se evaluó en pacientes con diversos grados de insuficiencia hepática (leve, moderada y grave) en comparación con pacientes con función hepática normal en un estudio de dosis única de 0.5 mg de semaglutida. Prediabetes y diabetes: La prediabetes y diabetes no tuvieron ningún efecto clínicamente relevante sobre la exposición de semaglutida. Inmunogenicidad: El desarrollo de anticuerpos antisemaglutida cuando se recibe tratamiento con semaglutida se produjo con poca frecuencia y la respuesta no pareció influir en la farmacocinética de semaglutida. Población pediátrica: Las propiedades farmacocinéticas de semaglutida se evaluaron en ensayos clínicos en pacientes adolescentes con obesidad o sobrepeso y al menos 1 comorbilidad asociada con el peso con edades entre los 12 y los 18 años (124 pacientes, peso corporal entre 61.6-211.9 kg). La exposición a semaglutida en adolescentes fue similar a la de los adultos con obesidad o sobrepeso. No se ha estudiado la seguridad y la eficacia de semaglutida en niños menores de 12 años. Datos preclínicos de seguridad: Los datos de los ensayos preclínicos no muestran riesgos especiales para los seres humanos según los ensayos convencionales de seguridad farmacológica, toxicidad a dosis repetidas o genotoxicidad. Los tumores no letales de células C de tiroides observados en roedores son un efecto de clase de los agonistas del receptor de GLP-1. Según los ensayos de carcinogenicidad de 2 años realizados en ratas y ratones, semaglutida causó tumores de células C de tiroides a exposiciones clínicamente significativas. No se observó ningún otro tumor relacionado con el tratamiento. Los tumores de células C observados en roedores están provocados por un mecanismo especifico no genotóxico mediado por el receptor de GLP-1 al que los roedores son especialmente sensibles. La relevancia en humanos se considera baja, pero no se puede excluir completamente. En estudios de fertilidad realizados en ratas, semaglutida no afectó a la conducta de apareamiento ni a la fertilidad de los machos. En las ratas hembra, se observó un aumento de la duración del ciclo estral y una ligera disminución de los cuerpos lúteos (ovulaciones) en dosis asociadas con pérdida de peso corporal materno. En los estudios de desarrollo embriofetal realizados en ratas, semaglutida causó embriotoxicidad por debajo de exposiciones clínicamente significativas. semaglutida provocó disminuciones pronunciadas del peso corporal materno y reducciones en términos de supervivencia y crecimiento embrionarios. En los fetos, se observaron importantes malformaciones esqueléticas y viscerales, con afectación de huesos largos, costillas, vértebras, cola, vasos sanguíneos y ventrículos cerebrales. Las evaluaciones mecanísticas realizadas indicaron que la embriotoxicidad estaba relacionada con una alteración del suministro de nutrientes al embrión a través del saco vitelino de la rata, mediada por el receptor de GLP-1. Debido a las diferencias entre especies en términos de anatomía y función del saco vitelino y a la falta de expresión del receptor de GLP-1 en el saco vitelino de primates no humanos, se considera que es improbable que este mecanismo sea relevante en humanos. Sin embargo, no se puede excluir un efecto directo de semaglutida en el feto. En los estudios de toxicidad para el desarrollo realizados en conejos y monos cynomolgus, se observó un aumento del número de casos de interrupción de la gestación y un ligero aumento de la incidencia de anomalías fetales a exposiciones clínicamente significativas. Estos hallazgos coincidieron con una marcada pérdida de peso corporal materna de hasta el 16 %. Se desconoce si estos efectos están relacionados con la reducción de la ingesta alimentaria materna como efecto directo del GLP-1. El crecimiento y el desarrollo posnatales se evaluaron en monos cynomolgus. Las crías fueron ligeramente más pequeñas al nacer, pero se recuperaron durante el periodo de lactancia. Semaglutida causó un retraso de la madurez sexual en ratas jóvenes, tanto en machos como en hembras. Estos retrasos no afectaron a la fertilidad y a la capacidad reproductora de ambos sexos o a la capacidad de las hembras para mantener la gestación. CONSERVACION: Conservar en heladera, entre 2 ºC a 8 ºC. No congelar. Descartar una vez utilizado. OBSERVACIONES: Este medicamento debe ser usado exclusivamente bajo prescripción y vigilancia médica y no puede repetirse sin nueva receta médica. Mantener este medicamento fuera del alcance de los niños. Conservar en su envase original. No utilice este medicamento después de la fecha de caducidad que aparece en el estuche. La fecha de caducidad es el último día del mes que se indica. Ante cualquier inconveniente con el producto el paciente puede contactarse al centro de atención al cliente de Laboratorio Elea 0800 333 3532. O bien llenar la ficha que está en la página web de la ANMAT: http://www.anmat.gov.ar/farmacovigilacia/Notificar.asp o llamar a la ANMAT responde 0800 333 1234. PRESENTACIONES: Obetide 0.25 mg/dosis: Envase conteniendo 2/4/8 jeringas prellenadas de 0.2 ml + 2/4/8 agujas descartables + 2/4/8 toallitas descartables saturadas en alcohol isopropílico al 70 %. Obetide 0.5 mg/dosis: Envase conteniendo 2/4/8 jeringas prellenadas de 0.4 ml + 2/4/8 agujas descartables + 2/4/8 toallitas descartables saturadas en alcohol isopropílico al 70 %. Obetide 1 mg/dosis: Envase conteniendo 2/4/8 jeringas prellenadas de 0.8 ml + 2/4/8 agujas descartables + 2/4/8 toallitas descartables saturadas en alcohol isopropílico al 70 %. Obetide 1.7 mg/dosis: Envase conteniendo 2/4/8 jeringas prellenadas de 0.53 ml + 2/4/8 agujas descartables + 2/4/8 toallitas descartables saturadas en alcohol isopropílico al 70 %. Obetide 2.4 mg/dosis: Envase conteniendo 2/4/8 jeringas prellenadas de 0.75 ml + 2/4/8 agujas descartables + 2/4/8 toallitas descartables saturadas en alcohol isopropílico al 70 %. |
||


{kind=link}
{kind=link}
 Software para farmacias |
 Vademécum Digital |
| El contenido de este sitio está protegido por el derecho de propiedad intelectual, encontrándose prohibida su reproducción y distribución total o parcial por el medio que sea. | |