Composición: Cada comprimido contiene: 10 mg de Rosuvastatina (como sal cálcica) y 10 mg de Ezetimiba. Excipientes c.s.: Lactosa Monohidrato, Celulosa Microcristalina, Croscarmelosa Sódica, Crospovidona, Estearato de Magnesio, Povidona, Lauril Sulfato de Sodio.
Indicaciones:Hipercolesterolemia primaria: Rovartal Plus está indicado como adyuvante de la dieta para el tratamiento de la hipercolesterolemia primaria, como terapia de sustitución en pacientes adultos adecuadamente controlados con los productos individuales, dados simultáneamente en el mismo nivel de dosis que en la combinación de dosis fija, pero como productos separados. Prevención de eventos cardiovasculares: Rovartal Plus está indicada para reducir los riesgos de eventos cardiovasculares como terapia de sustitución en pacientes con enfermedad coronaria e historial de síndrome coronario agudo, que se hayan mantenido bajo control adecuadamente utilizando los mismos principios activos y dosis, pero de forma separada que los incluidos en la mezcla a dosis fija, pero en medicamentos separados.
Propiedades:Propiedades farmacodinámicas: Rosuvastatina: Mecanismo de acción: La rosuvastatina es un inhibidor selectivo y competitivo de la HMG-CoA reductasa, la enzima limitante que convierte la 3-hidroxi-3-metilglutaril coenzima A en mevalonato, un precursor del colesterol. La rosuvastatina actúa principalmente en el hígado, que es el órgano diana para reducir el colesterol. La rosuvastatina aumenta el número de receptores de LDL hepáticos en la superficie celular, lo que aumenta la absorción y el catabolismo de la LDL, además de inhibir la síntesis hepática de VLDL. Esto reduce el número total de partículas de VLDL y LDL.
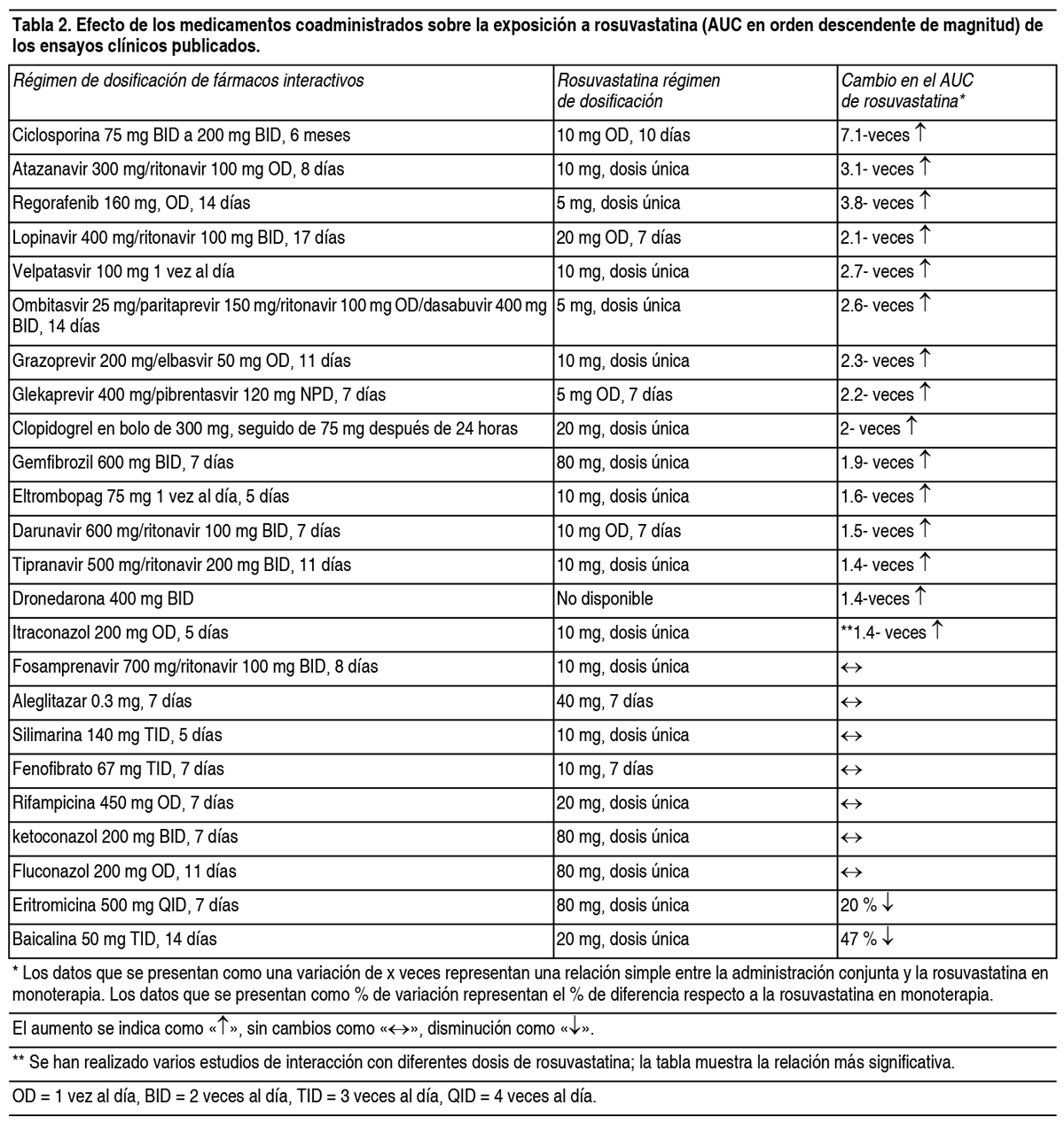
Efectos farmacodinámicos: La rosuvastatina reduce el colesterol LDL elevado, el colesterol total y los triglicéridos y aumenta el colesterol HDL. También disminuye los valores de ApoB, C-noHDL, C-VLDL, TG-VLDL e incrementa los valores de ApoA1 (ver Tabla 1). La rosuvastatina también disminuye los cocientes de C-LDL/C-HDL, C-total/C-HDL, C-noHDL/C-HDL y ApoB/ApoA1.
El efecto terapéutico se logra en 1 semana desde el inicio del tratamiento y el 90 % de la respuesta máxima se logra en 2 semanas. La respuesta máxima generalmente se logra en 4 semanas y se mantiene a partir de entonces. Ezetimiba: Mecanismo de acción: La ezetimiba inhibe la absorción de colesterol y esteroles vegetales relacionados en el intestino delgado. La ezetimiba es activo por vía oral y tiene un mecanismo de acción que difiere de otras clases de agentes reductores del colesterol (como estatinas, agentes secretores de ácidos biliares [resinas], derivados del ácido fíbrico y estanoles vegetales). El objetivo molecular de la ezetimiba es el transportador de esterol Niemann-Pick C1-Like 1 (NPC1L1), que es responsable de la absorción de colesterol y fitoesteroles en el intestino. La ezetimiba se posiciona en las microvellosidades del intestino delgado e inhibe la absorción de colesterol, reduciendo así la cantidad de colesterol que se suministra al hígado desde el intestino. En un ensayo clínico de 2 semanas de 18 pacientes con hipercolesterolemia, la ezetimiba inhibió la absorción intestinal de colesterol en un 54 % en comparación con el placebo. Se han realizado varios estudios preclínicos para determinar la selectividad de la ezetimiba con respecto a la inhibición de la absorción de colesterol. La ezetimiba inhibió la absorción de [14 C]-colesterol sin afectar la absorción de triglicéridos, ácidos grasos, ácidos biliares, progesterona, etinilestradiol o las vitaminas liposolubles A y D. Las estatinas reducen la síntesis de colesterol en el hígado y, juntos, estos diferentes mecanismos se complementan y contribuyen a la reducción del colesterol. Eficacia clínica rosuvastatina/ezetimiba en combinación: En un estudio clínico de 6 semanas, aleatorizado, doble ciego, de grupos paralelos, se evaluó la seguridad y eficacia de la ezetimiba (10 mg) ańadida al tratamiento estable con rosuvastatina en comparación con la titulación de rosuvastatina de 5 a 10 mg o de 10 a 20 mg. La población del estudio incluyó a 440 pacientes con riesgo moderado a alto de enfermedad coronaria con niveles de colesterol LDL más altos que los recomendados en el Panel III de Tratamiento para Adultos del Programa Nacional de Educación sobre el Colesterol (<100 mg/dl para pacientes de riesgo moderado/alto sin enfermedad vascular aterosclerótica o <70 mg/dl para pacientes de alto riesgo con enfermedad vascular aterosclerótica). Los resultados generales mostraron que la ezetimiba ańadida al tratamiento estable con rosuvastatina 5 mg o 10 mg produjo una reducción adicional del colesterol LDL de un 21 %. Por el contrario, duplicar la rosuvastatina a 10 mg o 20 mg redujo el colesterol LDL en un 5.7 %. En comparación con una titulación para duplicar la dosis de rosuvastatina, ezetimiba 10 mg ańadidos al tratamiento estable de rosuvastatina de 5 mg o 10 mg condujeron a mayores reducciones en muchos parámetros lipídicos, permitiendo que un porcentaje significativamente mayor de pacientes lograra los niveles de c-LDL meta, según su riesgo CDV. Propiedades farmacocinéticas: No se ha observado ninguna interacción farmacocinética clínicamente significativa entre los dos componentes de Rovartal Plus. Los valores medios de AUC y Cmáx para ezetimiba total y rosuvastatina no fueron diferentes entre los grupos de monoterapia y de combinación con rosuvastatina 10 mg y ezetimiba 10 mg. Absorción:
Ezetimiba: Después de la administración oral, la ezetimiba se absorbe rápidamente y se conjuga en gran medida con un fenol glucurónido farmacológicamente activo (ezetimiba glucurónido). Las concentraciones plasmáticas máximas medias (Cmáx) se alcanzan en 1 a 2 horas para el glucurónido de ezetimiba y de 4 a 12 horas para ezetimiba. No se puede determinar la biodisponibilidad absoluta de ezetimiba, ya que el compuesto es prácticamente insoluble en solución acuosa para inyección. La coadministración de alimentos (dieta rica en grasas o baja en grasas) no tuvo ningún efecto sobre la biodisponibilidad oral de ezetimiba administrada en comprimidos de 10 mg. La ezetimiba se puede administrar con o sin alimentos. Rosuvastatina: La concentración plasmática máxima se alcanza aprox. 5 horas después de la administración oral. La biodisponibilidad absoluta es de aproximadamente 20 %. Distribución: Ezetimiba: La ezetimiba y el glucurónido de ezetimiba se unen en un 99.7 % y de un 88 a un 92 % a las proteínas plasmáticas humanas, respectivamente. Rosuvastatina: La rosuvastatina es absorbida principalmente por el hígado, que es el sitio de acción principal para la síntesis de colesterol y la eliminación del colesterol LDL. El volumen de distribución es de aprox. 134 litros. Cerca del 90 % de la rosuvastatina se une a las proteínas plasmáticas, principalmente a la albúmina. Biotransformación: Ezetimiba: La ezetimiba se metaboliza principalmente en el intestino delgado y el hígado a través de la conjugación con glucurónidos (reacción de fase II) seguida de la excreción biliar. Se ha observado un metabolismo oxidativo mínimo (reacción de fase I) en todas las especies animales estudiadas. La ezetimiba y el glucurónido de ezetimiba son los principales compuestos derivados del fármaco detectados en el plasma y representan aproximadamente del 10 al 20 % y del 80 al 90 % del principio activo plasmático total, respectivamente. Tanto la ezetimiba como el glucurónido de ezetimiba se eliminan lentamente del plasma con evidencia de una recirculación enterohepática significativa. La vida media de la ezetimiba y el glucurónido de ezetimiba es de aproximadamente 22 horas. Rosuvastatina: La rosuvastatina se metaboliza de forma limitada (aproximadamente un 10 %). Los estudios de metabolismo in vitro con hepatocitos humanos indican que la rosuvastatina es, en pequeńa medida, sustrato del citocromo P450. La principal isoenzima implicada es la CYP2C9 y, en menor medida, la 2C19, 3A4 y la 2D6. Los principales metabolitos identificados son el metabolito N-desmetil y el lactónico. El metabolito N-desmetilo es aprox. 50 % menos activo que la rosuvastatina, mientras que el lactónico se considera clínicamente inactivo. La rosuvastatina representa más del 90 % de la actividad inhibidora de la HMG-CoA reductasa circulante. Eliminación: Ezetimiba: Tras la administración oral de 14C-ezetimiba (20 mg) a seres humanos, la ezetimiba total representó aproximadamente el 93 % de la radiactividad plasmática total. Aproximadamente el 78 % y el 11 % de la radiactividad administrada se recuperó en las heces y la orina, respectivamente, durante un período de recogida de 10 días. Después de 48 horas, no hubo niveles detectables de radiactividad plasmática. Rosuvastatina: Cerca del 90 % de la dosis de rosuvastatina se excreta inalterada en las heces (que consisten en principio activo absorbido y no absorbido) y el resto se excreta en la orina. Cerca del 5 % se excreta inalterado por la orina. La vida media de eliminación plasmática es de aprox. 19 horas. La vida media de eliminación no aumenta con dosis más altas. El aclaramiento plasmático medio geométrico es de aprox. 50 litros/hora (coeficiente de variación 21.7 %). Al igual que con otros inhibidores de la HMG-CoA reductasa, la proteína de transporte de membrana OATP-C participa en la absorción de rosuvastatina en el hígado. Esta proteína de transporte es importante en la eliminación hepática de la rosuvastatina. Linealidad: La exposición sistémica a la rosuvastatina aumenta en proporción a la dosis. No hay cambios en los parámetros farmacocinéticos después de dosis diarias repetidas. Poblaciones especiales: Insuficiencia hepática: Ezetimiba: Después de una dosis única de 10 mg de ezetimiba, el AUC medio de ezetimiba total aumentó aproximadamente 1.7 veces en pacientes con insuficiencia hepática leve (puntuación de Child-Pugh 5 o 6) en comparación con sujetos sanos. En un estudio de dosis repetidas de 14 días (10 mg al día), en pacientes con insuficiencia hepática moderada (puntuación de Child-Pugh de 7 a 9), el AUC media de ezetimiba total aumentó aproximadamente 4 veces el día 1 y el día 14 en comparación con los sujetos sanos. No se requiere un ajuste de dosis en pacientes con insuficiencia hepática leve. No se recomienda ezetimiba en pacientes con insuficiencia hepática moderada o grave (puntuación de Child-Pugh >9) debido al efecto desconocido del aumento de la exposición a ezetimiba en estos pacientes. Rosuvastatina: En un estudio en pacientes con diversos grados de insuficiencia hepática, no se observó un aumento de la exposición a rosuvastatina en pacientes con una puntuación Child-Pugh de 7 o menos. Sin embargo, se observó una exposición sistémica al menos 2 veces mayor en 2 pacientes con una puntuación de Child-Pugh de 8 y 9 en comparación con los pacientes con una puntuación de Child-Pugh más baja. No hay datos en pacientes con puntuaciones de Child-Pugh superiores a 9. Insuficiencia renal: Ezetimiba: Después de una dosis única de ezetimiba 10 mg en pacientes con enfermedad renal grave (n = 8; CrCl medio ≥30 ml/min), el AUC medio de ezetimiba total aumentó aproximadamente 1.5 veces en comparación con los sujetos sanos (n = 9). Este resultado no se considera clínicamente significativo. No se requiere un ajuste de dosis para pacientes con insuficiencia renal. Otro paciente de este estudio (postrasplante renal y que recibió varios fármacos diferentes, incluida ciclosporina) tuvo un aumento de 12 veces en la exposición total a ezetimiba. Rosuvastatina: En un estudio en pacientes con diversos grados de insuficiencia renal, la insuficiencia renal leve a moderada no tuvo ningún efecto sobre las concentraciones plasmáticas de rosuvastatina o del metabolito N-desmetilo. Sin embargo, los pacientes con insuficiencia renal grave (aclaramiento de creatinina <30 ml/min) tuvieron un aumento de 3 veces en la concentración plasmática y un aumento de 9 veces en la concentración del metabolito N-desmetilo en comparación con voluntarios sanos. Las concentraciones plasmáticas en estado estacionario de rosuvastatina en pacientes sometidos a hemodiálisis fueron de aprox. 50 % más grande en comparación con voluntarios sanos. Edad y género: Ezetimiba: La concentración plasmática de ezetimiba total es aproximadamente 2 veces mayor en los ancianos (≥65 ańos) que en los jóvenes (18 a 45 ańos). La reducción del colesterol LDL y el perfil de seguridad son comparables entre las personas mayores y las jóvenes tratadas con ezetimiba. No se requiere ajuste de dosis en ancianos. La concentración plasmática de ezetimiba total es ligeramente superior (aproximadamente un 20 %) en mujeres que en hombres. La reducción del colesterol LDL y el perfil de seguridad son comparables entre mujeres y hombres tratados con ezetimiba. Por tanto, no es necesario ajustar la dosis en función del sexo. Rosuvastatina: No se han observado efectos clínicamente relevantes sobre la farmacocinética de rosuvastatina con respecto a la edad y el sexo en adultos. Población pediátrica: Ezetimiba: La farmacocinética de la ezetimiba es similar entre nińos ≥6 ańos y adultos. No se dispone de datos farmacocinéticos en la población pediátrica <6 ańos. La experiencia clínica en nińos y adolescentes incluye pacientes con HoFH, HeFH o sitosterolemia. Rosuvastatina: 2 estudios farmacocinéticos con rosuvastatina (administrada en comprimidos) en pacientes pediátricos con hipercolesterolemia familiar heterocigótica de 10 a 17 o de 6 a 17 ańos (214 pacientes en total) mostraron que la exposición en pacientes pediátricos resultó ser comparable o menor que en pacientes adultos. Se encontró que la exposición a rosuvastatina era predecible en dosis y tiempo durante un período de 2 ańos. Etnicidad: Rosuvastatina: Los estudios de farmacocinética muestran aproximadamente una duplicación de la mediana del AUC y la Cmáx en los asiáticos (japoneses, chinos, filipinos, vietnamitas y coreanos) en comparación con los caucásicos. Los pacientes indo-asiáticos presentan un aumento de 1.3 veces en el AUC medio y la Cmáx. Un análisis farmacocinético poblacional no mostró diferencias clínicamente relevantes en la farmacocinética entre los grupos de raza blanca y negra. Polimorfismo genético: Rosuvastatina: El uso de inhibidores de la HMG-CoA reductasa, incluida la rosuvastatina, implica proteínas de transporte OATP1B1 y BCRP. En pacientes con polimorfismo genético en SLCO1B1 (OATP1B1) y/o ABCG2 (BCRP), existe el riesgo de una mayor exposición a rosuvastatina. Los polimorfismos individuales en SLCO1B1 c.521CC y ABCG2 c.421AA están asociados con una mayor exposición a rosuvastatina (AUC) en comparación con los genotipos SLCO1B1 c.521CC o ABCG2 c.421CC. Esta genotipificación específica no se ha establecido en la práctica clínica, pero para los pacientes con conocimiento de este tipo de polimorfismos se recomienda una dosis diaria más baja de rosuvastatina.
Posología: Rovartal Plus está indicado en pacientes adultos cuya hipercolesterolemia esté adecuadamente controlada mediante medicamentos monocomponentes administrados por separado a las mismas dosis que la combinación recomendada. El paciente debe mantener una dieta baja en grasas adecuada y debe seguir en esta dieta durante el tratamiento con este medicamento. La dosis diaria recomendada es de 1 comprimido de la concentración dada, con o sin comida. Este medicamento no es adecuado para el tratamiento inicial. El inicio del tratamiento o el ajuste de la dosis, si fuera necesario, solo se debe efectuar con los monocomponentes y, una vez establecidas las dosis correspondientes es posible cambiar a la combinación de dosis fija de la concentración adecuada. Rovartal Plus 10/10 comprimidos no es adecuado para el tratamiento de pacientes que requieran una dosis de 40 mg de rosuvastatina. La administración de este medicamento debe producirse ≥2 horas antes o ≥4 horas después de la administración de un secuestrante de ácidos biliares. Poblaciones especiales: Pacientes ancianos: Se recomienda una dosis inicial de 5 mg de rosuvastatina en pacientes >70 ańos. La combinación no es adecuada como tratamiento inicial. El inicio del tratamiento o, si es necesario, el ajuste de la dosis solo debe realizarse con los componentes individuales. Una vez que se han determinado las dosis correctas, se puede cambiar a la combinación de dosis fija adecuada. Insuficiencia hepática: No se requiere un ajuste de dosis en pacientes con insuficiencia hepática leve (puntuación de Child-Pugh de 5 a 6). No se recomienda el tratamiento con Rovartal Plus en pacientes con insuficiencia hepática moderada (puntuación de Child-Pugh de 7 a 9) o grave (puntuación de Child-Pugh >9). Rovartal Plus está contraindicado en pacientes con enfermedad hepática activa. Insuficiencia renal: No se requiere ajuste de dosis en pacientes con insuficiencia renal leve. La dosis inicial recomendada es de 5 mg de rosuvastatina en pacientes con insuficiencia renal moderada (aclaramiento de creatinina <60 ml/min). Una dosis de rosuvastatina 40 mg/ezetimiba 10 mg está contraindicada en pacientes con insuficiencia renal moderada. El uso de Rovartal Plus en pacientes con insuficiencia renal grave está contraindicado en todas las dosis. Etnicidad: Se ha observado un aumento de la exposición sistémica en asiáticos. La dosis inicial recomendada es de 5 mg de rosuvastatina para pacientes de origen asiático. La combinación de dosis fija no es adecuada para el tratamiento inicial. El inicio del tratamiento o el ajuste de la dosis debe realizarse con los componentes individuales. Una dosis de rosuvastatina 40 mg/ezetimiba 10 mg está contraindicada en estos pacientes. Polimorfismo genético: Se sabe que tipos específicos de polimorfismo genético pueden conducir a un aumento de la exposición a rosuvastatina. Se recomienda una dosis diaria más baja de rosuvastatina/ezetimiba cuando se sabe que los pacientes tienen tipos específicos de polimorfismo. Pacientes con factores predisponentes a la miopatía: La dosis inicial recomendada es de 5 mg de rosuvastatina para pacientes con factores que predisponen a la miopatía. La combinación de dosis fija no es adecuada para el tratamiento inicial. El inicio del tratamiento o el ajuste de la dosis debe realizarse con los componentes individuales. Una dosis de rosuvastatina 40 mg/ezetimiba 10 mg está contraindicada en algunos de estos pacientes. Población pediátrica: No se ha establecido todavía la seguridad y eficacia de Rovartal Plus en nińos menores de 18 ańos. No se pueden dar recomendaciones posológicas. Forma de administración: Rovartal Plus es para uso oral. Rovartal Plus se puede administrar en cualquier momento del día, con o sin comida. Los comprimidos deben tragarse enteros con un vaso de agua.
Efectos Colaterales:Resumen del perfil de seguridad: Las reacciones adversas informadas previamente con uno de los componentes individuales (ezetimiba o rosuvastatina) pueden ser reacciones adversas potenciales con Rovartal Plus. En ensayos clínicos de hasta 112 semanas de duración, se administró 10 mg de ezetimiba al día en monoterapia a 2396 pacientes o con una estatina a 11 308 pacientes o con fenofibrato a 185 pacientes. Los efectos secundarios fueron generalmente leves y transitorios. La incidencia global de reacciones adversas fue del mismo nivel para ezetimiba que para placebo. De forma similar, la proporción de pacientes que interrumpieron el estudio debido a reacciones adversas fue comparable a la de ezetimiba y placebo. Las reacciones adversas de la rosuvastatina suelen ser leves y transitorias. En ensayos clínicos controlados, menos del 4 % de los pacientes tratados con rosuvastatina interrumpieron los estudios debido a reacciones adversas. Tabla de reacciones adversas: Las tasas de reacciones adversas se clasifican de la siguiente manera: Muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a <1/10), poco frecuentes (≥ 1/1000 a <1/100), raras (≥ 1/10 000 a <1/1000), muy raras (<1/10 000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Al igual que con otros inhibidores de la HMG-CoA reductasa, la incidencia de reacciones adversas parece depender de la dosis. Efecto sobre la función renal: En pacientes tratados con rosuvastatina, se observó proteinuria, principalmente de origen tubular, en la prueba con tira reactiva. Se observaron cambios en la proteinuria desde nada o trazas hasta un resultado ++ o superior en <1 % de los pacientes en algún momento del tratamiento con 10 y 20 mg respectivamente y aproximadamente en el 3 % de los pacientes tratados con 40 mg. Con la dosis de 20 mg, se observó un menor incremento en el cambio desde nada o trazas a +. En la mayoría de los casos, la proteinuria disminuyó o desapareció espontáneamente con el tratamiento continuado. La revisión de los ensayos clínicos y la experiencia clínica posterior a la comercialización no han demostrado hasta ahora una relación causal entre la proteinuria y la enfermedad renal aguda o progresiva. Se ha observado hematuria en pacientes tratados con rosuvastatina, pero los datos de los ensayos clínicos muestran una baja incidencia. Efecto sobre el músculo esquelético: Se han registrado efectos sobre el músculo esquelético, por ej., mialgia, miopatía (incluyendo miositis) y, en casos raros, rabdomiólisis con o sin insuficiencia renal aguda, en sujetos tratados con rosuvastatina, independientemente de la dosis, pero especialmente con dosis >20 mg. Se ha observado un aumento relacionado con la dosis en los niveles de CK en pacientes que toman rosuvastatina. La mayoría de los casos fueron leves, asintomáticos y transitorios. Si los niveles de CK están elevados (>5 x LSN), se debe interrumpir el tratamiento. Efectos hepáticos: Al igual que con otros inhibidores de la HMG-CoA reductasa, se ha observado un aumento de las transaminasas relacionado con la dosis en un pequeńo número de pacientes tratados con rosuvastatina; la mayoría de los casos fueron leves, asintomáticos y transitorios. La frecuencia de notificación de rabdomiólisis, insuficiencia renal grave e insuficiencia hepática grave (principalmente en forma de aumento de las transaminasas hepáticas) es mayor con la dosis de 40 mg. Se han informado los siguientes efectos secundarios con algunas estatinas: Disfunción sexual. Casos poco frecuentes de enfermedad pulmonar intersticial, especialmente con tratamiento a largo plazo. Valores de laboratorio: En ensayos clínicos controlados con monoterapia, la incidencia de aumentos clínicamente significativos en las transaminasas séricas (ALT y/o AST ≥3 x LSN, consecutivos) fue la misma para ezetimiba (0.5 %) y placebo (0.3 %). En los estudios de terapia combinada, la incidencia fue del 1.3 % para los pacientes tratados con ezetimiba y una estatina de forma concomitante y del 0.4 % para los pacientes tratados con una estatina sola. Estos aumentos fueron normalmente asintomáticos, no se asociaron con colestasis y volvieron a los valores iniciales después de la interrupción del tratamiento o al continuarlo. En los ensayos clínicos, se notificó CK >10 x LSN en 4 de 1674 (0.2 %) pacientes que recibieron ezetimiba sola, en comparación con 1 de cada 786 (0.1 %) pacientes que recibieron placebo y 1 de cada 917 (0.1 %) pacientes que recibieron ezetimiba concomitantemente con una estatina, en comparación con 4 de 929 (0.4 %) pacientes que recibieron una estatina sola. No hubo un mayor grado de miopatía y rabdomiólisis asociado con ezetimiba en comparación con los grupos de control correspondientes (placebo o estatina sola). Población pediátrica: No se ha establecido todavía la seguridad y eficacia de Rovartal Plus en nińos menores de 18 ańos. Rosuvastatina: En un ensayo clínico de 52 semanas de duración de tratamiento, realizado en nińos y adolescentes, se observó un incremento en los niveles de CK >0xLSN y aumento de los síntomas musculares después del ejercicio o actividad física, con mayor frecuencia en
comparación con adultos. En otros aspectos, el perfil de seguridad de la rosuvastatina fue similar en nińos y adolescentes en comparación con adultos. Ezetimiba: En un estudio en pacientes pediátricos (de 6 a 10 ańos de edad) con hipercolesterolemia familiar heterocigota o hipercolesterolemia no familiar (n = 138), se observó elevaciones de ALT y/o AST (≥3 x LSN, consecutivas) en 1.1 % (1 paciente) de los pacientes del grupo de ezetimiba en comparación con el 0 % del grupo de placebo. No hubo aumento de CK (≥10 x LSN). No se informaron casos de miopatía. En un estudio separado en adolescentes (10 a 17 ańos) con hipercolesterolemia familiar heterocigótica (n = 248), se observó elevaciones de ALT y/o AST (≥3 x LSN, consecutivas) en el 3 % (4 pacientes) de los pacientes en el grupo de ezetimiba/simvastatina en comparación con el 2 % (2 pacientes) en el grupo de monoterapia con simvastatina. Para el aumento de CK (≥10 x LSN), las cifras fueron 2 % (2 pacientes) y 0 %, respectivamente. No se informaron casos de miopatía.
Contraindicaciones: Hipersensibilidad a los principios activos o a alguno de los excipientes de este medicamento. Durante el embarazo, la lactancia y en mujeres en edad fértil que no utilicen métodos anticonceptivos adecuados. Enfermedad hepática activa o aumentos persistentes e inexplicables de las transaminasas séricas y cualquier aumento de las transaminasas séricas que supere 3 veces el límite superior de lo normal (LSN). Insuficiencia renal grave (aclaramiento de creatinina <30 ml/min). Miopatía. Pacientes que reciben la combinación concomitante de sofosbuvir/velpatasvir/voxilaprevir. Tratamiento concomitante con ciclosporina.
Advertencias:Reacciones adversas cutáneas graves: Se han notificado reacciones adversas cutáneas graves con la rosuvastatina, incluyendo el síndrome de Stevens-Johnson (SJS, por sus siglas en inglés) y la reacción al fármaco con eosinofilia y síntomas sistémicos (DRESS, por sus siglas en inglés), que pueden poner en peligro la vida o resultar mortales. En el momento de la prescripción, se debe advertir a los pacientes de los signos y síntomas de las reacciones cutáneas graves y ser monitorizados estrechamente. Si aparecen signos y síntomas indicativos de esta reacción, se debe interrumpir Rovartal Plus inmediatamente y se debe considerar un tratamiento alternativo. Si el paciente ha desarrollado una reacción grave como SJS o DRESS con el uso de Rovartal Plus, el tratamiento con Rovartal Plus no se debe reiniciar en este paciente en ningún momento. Efecto sobre los músculos esqueléticos: Se han notificado efectos sobre el músculo esquelético, como mialgia, miopatía y, en raras ocasiones, rabdomiólisis, en pacientes tratados con rosuvastatina, independientemente de la dosis, pero especialmente con dosis >20 mg. Al igual que con otros inhibidores de la HMG-CoA reductasa, la experiencia poscomercialización de la rabdomiólisis es mayor para dosis de 40 mg. Tras la comercialización de ezetimiba, se han notificado casos de miopatía y rabdomiólisis. Sin embargo, se ha informado rabdomiólisis muy raramente cuando ezetimiba se toma solo y cuando ezetimiba se toma junto con otros agentes asociados con un mayor riesgo de rabdomiólisis. Si se sospecha miopatía en función de los síntomas musculares o se confirma por los niveles de creatinfosfocinasa (CK), los pacientes que toman Rovartal Plus y otros medicamentos concomitantes deben interrumpir el tratamiento inmediatamente. Todos los pacientes que inicien el tratamiento con Rovartal Plus deben ser informados del riesgo de miopatía y se les debe animar a que informen inmediatamente de dolor, sensibilidad o debilidad muscular inexplicable. Medición de creatina quinasa: La creatina quinasa (CK) no debe medirse después de un esfuerzo físico intenso o en presencia de otra causa alternativa plausible de CK elevada, ya que esto complica la interpretación de los resultados. Si la CK está significativamente elevada al inicio del estudio (>5 x LSN), se deben tomar nuevas mediciones dentro de los 5-7 días para confirmar los resultados. Si la segunda medición confirma un valor de CK basal >5 x LSN, no se debe iniciar el tratamiento. Antes del tratamiento: Se debe tener precaución en pacientes con factores predisponentes a miopatía/rabdomiólisis. Estos factores pueden ser: Insuficiencia renal. Hipotiroidismo. Enfermedad muscular previamente conocida en el paciente o enfermedad muscular hereditaria en la familia. Casos previos de toxicidad muscular con otros inhibidores de la HMG-CoA reductasa o fibratos. Abuso de alcohol. Edad >70 ańos. Situaciones en las que puede haber un aumento de los niveles plasmáticos. Tratamiento concomitante con fibratos. En estos pacientes, se debe sopesar el riesgo del tratamiento con el beneficio potencial y se recomienda la monitorización clínica. Si los niveles de CK están significativamente elevados al inicio del estudio (>5 x LSN), no debe iniciarse el tratamiento. Durante el tratamiento: Se debe pedir a los pacientes que informen inmediatamente de dolor muscular inexplicable, debilidad muscular o calambres musculares, especialmente si esto está asociado con malestar o fiebre. Los niveles de CK deben medirse en estos pacientes. El tratamiento debe suspenderse si los niveles de CK están significativamente elevados (>5 x LSN) o si los síntomas musculares son graves y causan malestar diario (incluso si los niveles de CK son ≤5 x LSN). Si los síntomas se resuelven y los niveles de CK vuelven a la normalidad, se puede considerar reiniciar el tratamiento con rosuvastatina u otro inhibidor de la HMG-CoA reductasa y realizar un seguimiento cuidadoso del paciente. No se requiere un control de rutina de los niveles de CK en pacientes asintomáticos. Se han notificado casos muy raros de miopatía necrotizante inmunomediada (IMNM) durante y después del tratamiento con estatinas, incluida la rosuvastatina. La IMNM se caracteriza clínicamente por debilidad muscular proximal y CK sérica elevada, que persiste a pesar de la interrupción del tratamiento con estatinas. En los ensayos clínicos no hubo evidencia de un aumento de los efectos musculoesqueléticos en el reducido número de pacientes tratados con rosuvastatina y tratamiento concomitante. Sin embargo, ha habido un aumento de miositis y miopatía en pacientes tratados con otros inhibidores de la HMG-CoA reductasa en combinación con fibratos, incluidos gemfibrozilo, ciclosporina, ácido nicotínico, antifúngicos azólicos, inhibidores de proteasa y antibióticos macrólidos. El gemfibrozil aumenta el riesgo de miopatía cuando se coadministra con ciertos inhibidores de la HMG-CoA reductasa. Por lo tanto, no se recomienda una combinación de rosuvastatina y gemfibrozil. El beneficio de un cambio adicional en los niveles de lípidos con el uso combinado de rosuvastatina con fibratos o niacina debe sopesarse cuidadosamente contra el riesgo potencial de tales combinaciones. Dosis de 40 mg de rosuvastatina está contraindicada con el uso concomitante de fibratos. Rovartal Plus no debe utilizarse en pacientes con una enfermedad aguda y grave que indique miopatía o que predisponga al desarrollo de insuficiencia renal secundaria a rabdomiólisis (por ejemplo, sepsis, hipotensión, cirugía mayor, traumatismo, alteraciones metabólicas, endocrinas y electrolíticas graves o convulsiones no controladas). Efectos hepáticos: En estudios controlados en los que los pacientes recibieron ezetimiba concomitantemente con una estatina, hubo un aumento consecuente de las transaminasas séricas (≥3 x LSN). Se recomienda realizar pruebas de función hepática antes y 3 meses después de iniciar el tratamiento. Se debe interrumpir el tratamiento con rosuvastatina o reducir la dosis si los niveles de transaminasas séricas exceden 3 veces el límite superior de lo normal. La frecuencia de notificación de insuficiencia hepática grave (principalmente en forma de aumento de las transaminasas hepáticas) cuando se utilizó después del lanzamiento fue mayor para la dosis de 40 mg. En pacientes con hipercolesterolemia secundaria causada por hipotiroidismo o síndrome nefrótico, la enfermedad subyacente debe tratarse antes de iniciar el tratamiento con Rovartal Plus. Debido a los efectos desconocidos del aumento de la exposición a ezetimiba en pacientes con insuficiencia hepática moderada o grave, no se recomienda este medicamento en estos pacientes. Enfermedad hepática y alcohol: Al igual que con otros inhibidores de la HMG-CoA reductasa, la rosuvastatina debe usarse con precaución en pacientes que consumen grandes cantidades de alcohol y/o tienen antecedentes de enfermedad hepática. Efecto sobre la función renal: En pacientes tratados con dosis altas de rosuvastatina, especialmente 40 mg, se observó proteinuria de origen predominantemente tubular en la prueba de tira de orina. En la mayoría de los casos, esto fue transitorio o periódico. No se ha demostrado que la proteinuria sea un signo de enfermedad renal aguda o progresiva. Después del lanzamiento, la frecuencia de notificación de eventos renales graves ha sido mayor para dosis de 40 mg. Para pacientes tratados con 40 mg de rosuvastatina, se debe considerar una evaluación de la función renal en el seguimiento rutinario. Diabetes mellitus: Existe alguna evidencia que sugiere que las estatinas como clase aumentan la glucosa en sangre, por lo que, en algunos pacientes con alto riesgo de diabetes en el futuro, un cuidado convencional de la diabetes es apropiado. Sin embargo, este riesgo se compensa con la reducción del riesgo vascular con las estatinas y, por lo tanto, no debería ser una razón para interrumpir el tratamiento con estatinas. Los pacientes con riesgo (glucosa en ayunas de 5.6 a 6.9 mmol/l, IMC >30 kg/m2, aumento de triglicéridos, hipertensión) deben ser controlados tanto clínica como bioquímicamente de acuerdo con las directrices nacionales. En el estudio JUPITER, la incidencia global notificada de diabetes mellitus fue del 2.8 % con rosuvastatina y del 2.3 % con placebo, principalmente en pacientes con glucosa en ayunas de 5.6 a 6.9 mmol/l. Enfermedad pulmonar intersticial: Se han notificado casos específicos de enfermedad pulmonar intersticial con determinadas estatinas, especialmente con el tratamiento a largo plazo. Los síntomas pueden incluir disnea, tos seca y empeoramiento del estado general (fatiga, pérdida de peso y fiebre). Si se sospecha que un paciente ha desarrollado enfermedad pulmonar intersticial, se debe suspender el tratamiento con estatinas. Inhibidores de la proteasa: Se ha observado un aumento de la exposición sistémica a rosuvastatina en sujetos que reciben rosuvastatina de forma concomitante con varios inhibidores de la proteasa en combinación con ritonavir. Se debe considerar tanto el beneficio de la reducción de lípidos con Rovartal Plus en pacientes con VIH que reciben inhibidores de la proteasa como la posibilidad de que aumenten las concentraciones plasmáticas de rosuvastatina al iniciar y aumentar la dosis de rosuvastatina en pacientes tratados con inhibidores de la proteasa. No se recomienda el tratamiento concomitante con ciertos inhibidores de la proteasa a menos que se ajuste la dosis de rosuvastatina. Fibratos: No se ha establecido la seguridad y eficacia de ezetimiba coadministrado con fibratos. Si se sospecha de cálculos biliares en un paciente que recibe este medicamento y fenofibrato, se requiere un examen de la vesícula biliar y se debe suspender el tratamiento. Anticoagulantes: El índice internacional normalizado (INR) debe controlarse adecuadamente si Rovartal Plus se administra además de con warfarina, con otros anticoagulantes de tipo cumarina o fluindiona. Ácido fusídico: Rovartal Plus no debe usarse concomitantemente con formulaciones sistémicas de ácido fusídico o dentro de los 7 días posteriores al final del tratamiento con ácido fusídico. En pacientes en los que el uso de ácido fusídico sistémico se considera importante, se debe interrumpir el tratamiento con estatinas. Se ha notificado rabdomiólisis (incluidas algunas muertes) en pacientes que han utilizado una combinación de ácido fusídico y estatinas. Se debe advertir a los pacientes que busquen atención médica inmediata si experimentan síntomas de debilidad, dolor o sensibilidad muscular. La terapia con estatinas se puede reiniciar 7 días después de la última dosis de ácido fusídico. En circunstancias especiales donde se requiera el uso prolongado de ácido fusídico sistémico, p. ej., en el tratamiento de infecciones graves, se debe considerar la necesidad de un uso concomitante con Rovartal Plus en cada caso individual bajo una estrecha supervisión médica. Etnicidad: Los estudios farmacocinéticos muestran un aumento de la exposición en asiáticos en comparación con los caucásicos. Población pediátrica: No se recomienda el tratamiento con Rovartal Plus en nińos y adolescentes menores de 18 ańos debido a la escasez de datos sobre seguridad y eficacia. Información importante sobre alguno de los componentes de este medicamento: Este medicamento contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, deficiencia total de lactasa o problemas de absorción de glucosa o galactosa no deben tomar este medicamento. Este medicamento contiene menos de 1 mmol de sodio (23 mg) por comprimido y es esencialmente «exento de sodio».
Precauciones:Embarazo: No existen datos clínicos sobre el uso de ezetimiba durante el embarazo. Los estudios en animales con ezetimiba solo no han mostrado evidencia de efectos dańinos directos o indirectos con respecto al embarazo, desarrollo embriofetal, parto o desarrollo posnatal. Debido a que el colesterol y otros productos de la biosíntesis del colesterol son esenciales para el desarrollo fetal, el riesgo potencial de inhibición de la HMG-CoA reductasa supera los beneficios del tratamiento durante el embarazo. Los estudios en animales proporcionan pruebas limitadas de toxicidad para la reproducción. Durante el embarazo, el tratamiento con este medicamento debe interrumpirse inmediatamente. Lactancia: Los estudios en ratas han demostrado que la ezetimiba se excreta en la leche materna. No se sabe si la ezetimiba se excreta en la leche materna humana. La rosuvastatina se excreta en la leche de ratas. No hay datos sobre la excreción en la leche materna humana. Fertilidad: No hay datos de ensayos clínicos sobre el efecto de la ezetimiba o la rosuvastatina en la fertilidad humana. La ezetimiba no mostró ningún efecto sobre la fertilidad en ratas macho o hembra. A dosis más altas, la rosuvastatina mostró toxicidad testicular en monos y perros. Efectos sobre la habilidad para conducir o manejar maquinaria pesada: No se han realizado estudios sobre la capacidad para conducir y utilizar máquinas. Sin embargo, al conducir y utilizar máquinas, se debe tener en cuenta que se han notificado mareos.
Interacciones Medicamentosas:Combinaciones contraindicadas: Ciclosporina: La administración concomitante de Rovartal Plus con ciclosporina está contraindicada debido a la rosuvastatina. Durante el tratamiento concomitante con rosuvastatina y ciclosporina, los valores de AUC de rosuvastatina fueron en promedio 7 veces más altos que los observados en voluntarios sanos (ver Tabla 2). La coadministración no afectó las concentraciones plasmáticas de ciclosporina. En un estudio de 8 pacientes postrasplante de rińón con aclaramiento de creatinina >50 ml/min con una dosis estable de ciclosporina, una dosis única de 10 mg de ezetimiba resultó en un aumento de 3.4 veces (rango de 2.3 a 7.9 veces) en el AUC medio de ezetimiba total en comparación con un grupo de control sano, de otro estudio (n = 17) que recibió ezetimiba solo. En un estudio diferente, en un paciente trasplantado renal con insuficiencia renal grave que recibía ciclosporina y otros múltiples medicamentos, se demostró una exposición 12 veces mayor a la ezetimiba total comparada con controles concurrentes que estaban recibiendo solo ezetimiba. En un estudio cruzado de 2 períodos, 12 voluntarios sanos recibieron 20 mg de ezetimiba al día durante 8 días y una dosis única de 100 mg de ciclosporina el día 7. Esto resultó en un aumento medio del 15 % en el AUC de ciclosporina (intervalo del 10 % de descenso al 51 % de aumento) en comparación con una dosis única de 100 mg de ciclosporina sola. No se ha realizado un estudio controlado sobre el efecto de ezetimiba coadministrado con ciclosporina en pacientes con trasplante renal. Combinaciones no recomendadas Fibratos y otros medicamentos reductores de lípidos: En pacientes que toman fenofibrato y ezetimiba, los médicos deben ser conscientes del riesgo potencial de cálculos biliares y enfermedad de la vesícula biliar. Si se sospecha de cálculos biliares en pacientes que toman ezetimiba y fenofibrato, se debe examinar la vesícula biliar y suspender el tratamiento. El uso concomitante de fenofibrato o gemfibrozil aumentó moderadamente la concentración total de ezetimiba (aproximadamente 1.5 y 1.7 veces, respectivamente). No se ha estudiado el uso de ezetimiba con otros fibratos. Los fibratos pueden aumentar la excreción de colesterol en la bilis y esto puede provocar cálculos biliares. En estudios con animales, ezetimiba a veces aumentó el colesterol en la vesícula biliar, pero no en todas las especies. No se puede descartar el riesgo asociado a cálculos biliares con el uso terapéutico de ezetimiba. El tratamiento concomitante con rosuvastatina y gemfibrozilo dio como resultado un aumento del doble de la Cmáx y el AUC de rosuvastatina. Según los datos de los estudios de interacción específicos, no se esperan interacciones farmacocinéticas relevantes con fenofibrato, pero pueden producirse interacciones farmacodinámicas. El gemfibrozil, los fenofibratos, otros fibratos y las dosis reductoras de lípidos (>1 g/día equivalente) de niacina (ácido nicotínico) aumentan el riesgo de miopatía cuando se administran concomitantemente con inhibidores de la HMG-CoA reductasa, probablemente porque pueden causar miopatía cuando se administran solos. Dosis de 40 mg de rosuvastatina con 10 mg de ezetimiba está contraindicada con el uso concomitante de fibratos. Inhibidores de la proteasa: Aunque se desconoce el mecanismo de acción exacto de la interacción, el tratamiento concomitante con inhibidores de la proteasa puede aumentar significativamente la exposición a rosuvastatina (ver Tabla 2). El tratamiento concomitante con rosuvastatina 10 mg y un producto combinado con 2 inhibidores de la proteasa (300 mg de atazanavir/100 mg de ritonavir) en voluntarios sanos, en un estudio farmacocinético, se asoció con aprox. un aumento de 3 y 7 veces en el AUC y Cmáx de rosuvastatina. Se puede considerar el uso concomitante de rosuvastatina y ciertas combinaciones de inhibidores de la proteasa después de considerar cuidadosamente el ajuste de la dosis de rosuvastatina en base al aumento esperado en la exposición a rosuvastatina (ver Tabla 2). Inhibidores de proteínas de transporte: La rosuvastatina es un sustrato para ciertas proteínas de transporte, como OATP1B1, un transportador de captación en el hígado, y BCRP, un transportador de eflujo. La coadministración de rosuvastatina y medicamentos que inhiben estas proteínas transportadoras puede provocar un aumento de las concentraciones plasmáticas de rosuvastatina y un aumento del riesgo de miopatía (ver Tabla 2). Ácido fusídico: El riesgo de miopatía, incluida la rabdomiólisis, puede aumentar con la coadministración de ácido fusídico sistémico y estatinas. El mecanismo de esta interacción (si es farmacodinámico, farmacocinético o ambos) aún se desconoce. Se ha notificado rabdomiólisis (incluidos aquellos con desenlace fatal) en pacientes que recibieron esta combinación. Si se requiere tratamiento con ácido fusídico sistémico, se debe interrumpir el tratamiento con rosuvastatina durante todo el período de tratamiento con ácido fusídico. Otras interacciones: Enzimas del citocromo P450: Los resultados de estudios in vitro e in vivo muestran que la rosuvastatina no inhibe ni induce las isoenzimas del citocromo P450. Además, la rosuvastatina es un sustrato con poca afinidad para estas isoenzimas. Por lo tanto, no se esperan interacciones debido al metabolismo del citocromo P450. No se han observado interacciones clínicamente relevantes entre rosuvastatina y fluconazol (un inhibidor de CYP2C9 y CYP3A4) o ketoconazol (un inhibidor de CYP2A6 y CYP3A4). En los estudios preclínicos, se ha demostrado que ezetimiba no induce las enzimas metabolizadoras de fármacos del sistema del citocromo P450. No se han observado interacciones farmacocinéticas clínicamente importantes entre ezetimiba y fármacos metabolizados por las isoenzimas 1A2, 2D6, 2C8, 2C9 y 3A4 del citocromo P450 o por la N-acetiltransferasa. Antiácidos: La coadministración de antiácidos redujo la tasa de absorción de ezetimiba, pero no tuvo ningún efecto sobre su biodisponibilidad. Esta reducción de la tasa de absorción no se consideró clínicamente relevante. La coadministración de rosuvastatina y una suspensión antiácida que contiene hidróxido de aluminio y magnesio dio como resultado una reducción en la concentración plasmática de rosuvastatina de aproximadamente un 50 %. Este efecto se redujo cuando se administró el antiácido 2 horas después de la rosuvastatina. No se ha estudiado la importancia clínica de esta interacción. Colestiramina: La coadministración de colestiramina redujo el área media bajo la curva (AUC) de ezetimiba total (ezetimiba + glucurónido de ezetimiba) en aprox. 55 %. Por tanto, esta interacción puede reducir el aumento de la reducción del colesterol LDL debido a la adición de ezetimiba a la colestiramina. Anticoagulantes, antagonistas de la vitamina K: La administración concomitante de ezetimiba (10 mg 1 vez al día) no tuvo un efecto significativo sobre la biodisponibilidad de warfarina y el tiempo de protrombina en un estudio en 12 varones adultos sanos. Sin embargo, en la experiencia poscomercialización, se ha notificado un aumento del índice internacional normalizado (INR) en pacientes que reciben ezetimiba además de warfarina o fluindiona. Si se administra Rovartal Plus además de warfarina u otros anticoagulantes de tipo cumarina o fluindiona, se debe controlar adecuadamente el INR. Al igual que con otros inhibidores de la HMG-CoA reductasa, el inicio del tratamiento o la titulación de la dosis con rosuvastatina en pacientes tratados de forma concomitante con antagonistas de la vitamina K (p. ej., warfarina u otros anticoagulantes cumarínicos) puede provocar un aumento del índice internacional normalizado (INR). La interrupción del tratamiento o la disminución de la dosis de rosuvastatina puede resultar en una reducción del INR. En tales casos, se debe realizar un seguimiento adecuado del INR. Eritromicina: El uso concomitante de rosuvastatina y eritromicina resultó en una reducción del 20 % en el AUC y una reducción del 30 % en la Cmáx de la rosuvastatina. Esta interacción puede deberse a un aumento de la motilidad intestinal provocado por la eritromicina. Anticonceptivos orales/terapia de reemplazo hormonal (THS): El uso concomitante de rosuvastatina y un anticonceptivo oral resultó en un aumento en el AUC de etinilestradiol y norgestrel de 26 % y 34 %, respectivamente. Se debe tener en cuenta el aumento de los niveles plasmáticos al elegir una dosis de anticonceptivo. No se dispone de datos farmacocinéticos de mujeres que reciben concomitantemente rosuvastatina y terapia de reemplazo hormonal, por lo que no se puede descartar un efecto similar. Sin embargo, la combinación es ampliamente utilizada por mujeres en ensayos clínicos y fue bien tolerada. En los estudios de interacción clínica, ezetimiba no tuvo ningún efecto sobre la farmacocinética de los anticonceptivos orales (etinilestradiol y levonorgestrel). Otros medicamentos: De acuerdo a los resultados de estudios específicos de interacción, no se esperan interacciones importantes entre rosuvastatina y digoxina. En estudios de interacción clínica, la ezetimiba no tuvo ningún efecto sobre la farmacocinética de dapsona, dextrometorfano, digoxina, glipizida, tolbutamida o midazolam cuando se administraron concomitantemente. La cimetidina coadministrada con ezetimiba no tuvo ningún efecto sobre la biodisponibilidad de ezetimiba. Interacciones que requieren un ajuste de la dosis de rosuvastatina (ver también Tabla 2). Cuando sea necesario administrar rosuvastatina con otros medicamentos que se sabe que aumentan la exposición a rosuvastatina, se debe ajustar la dosis de rosuvastatina. La dosis máxima diaria de rosuvastatina debe ajustarse de modo que sea poco probable que la exposición esperada a rosuvastatina exceda la administrada por una dosis diaria de 40 mg de rosuvastatina tomada sin el fármaco que interactúe, p. ej., una dosis de 20 mg de rosuvastatina con gemfibrozil (aumento de 1.9 veces) y una dosis de 10 mg de rosuvastatina con la combinación de ritonavir/atazanavir (aumento de 3.1 veces).
Sobredosificación: En caso de sobredosis, se deben tomar medidas sintomáticas y de soporte. Ezetimiba: En los ensayos clínicos, la administración de ezetimiba 50 mg/día a 15 sujetos sanos durante un máximo de 14 días o 40 mg/día a 18 pacientes con hipercolesterolemia primaria durante un máximo de 56 días fue generalmente bien tolerada. En animales, no se observó toxicidad después de dosis orales únicas de 5.000 mg/kg de ezetimiba en ratas y ratones y 3.000 mg/kg en perros. Se han notificado algunos casos de sobredosis de ezetimiba. La mayoría no se ha asociado con efectos secundarios. Los efectos secundarios notificados no han sido graves. Rosuvastatina: Se debe controlar la función hepática y el nivel de CPK. Es poco probable que la hemodiálisis tenga algún efecto.
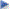 Laboratorio
Laboratorio