Composición:Alhemo® 15 mg/1.5 ml: 1 ml de solución contiene 10 mg de Concizumab*. Cada lapicera prellenada contiene 15 mg de Concizumab en 1.5 ml de solución (10 mg/ml). Alhemo® 60 mg/1.5 ml: 1 ml de solución contiene 40 mg de Concizumab*. Cada lapicera prellenada contiene 60 mg de concizumab en 1.5 ml de solución (40 mg/ml). Alhemo® 150 mg/1.5 ml: 1 ml de solución contiene 100 mg de Concizumab*. Cada lapicera prellenada contiene 150 mg de Concizumab en 1.5 ml de solución (100 mg/ml). Alhemo® 300 mg/3 ml: 1 ml de solución contiene 100 mg de Concizumab*. Cada lapicera prellenada contiene 300 mg de Concizumab en 3 ml de solución (100 mg/ml). *Concizumab es un anticuerpo monoclonal IgG4 humanizado, producido mediante tecnología de ADN recombinante en células de ovario de hámster chino (CHO). Excipientes: Clorhidrato de L-Arginina, L-Histidina, Cloruro de Sodio, Sacarosa, Polisorbato 80, Fenol, Ácido Clorhídrico e Hidróxido de Sodio (para ajuste del pH) y Agua para Inyectables. Forma farmacéutica: Solución inyectable en lapicera prellenada. Líquido transparente a ligeramente opalescente, incoloro a ligeramente amarillo y prácticamente libre de partículas visibles, que puede contener partículas de proteína traslúcidas a blancas. Solución isotónica de aproximadamente pH 6.
Acción Terapéutica:Grupo farmacoterapéutico: Antihemorrágicos, otros hemostáticos sistémicos. Código ATC: B02BX10.
Indicaciones:Alhemo® está indicado para la profilaxis de rutina de hemorragias en pacientes con: Hemofilia A (deficiencia congénita del factor VIII) con inhibidores del FVIII a partir de los 12 ańos de edad. Hemofilia B (deficiencia congénita del factor IX) con inhibidores del FIX a partir de los 12 ańos de edad.
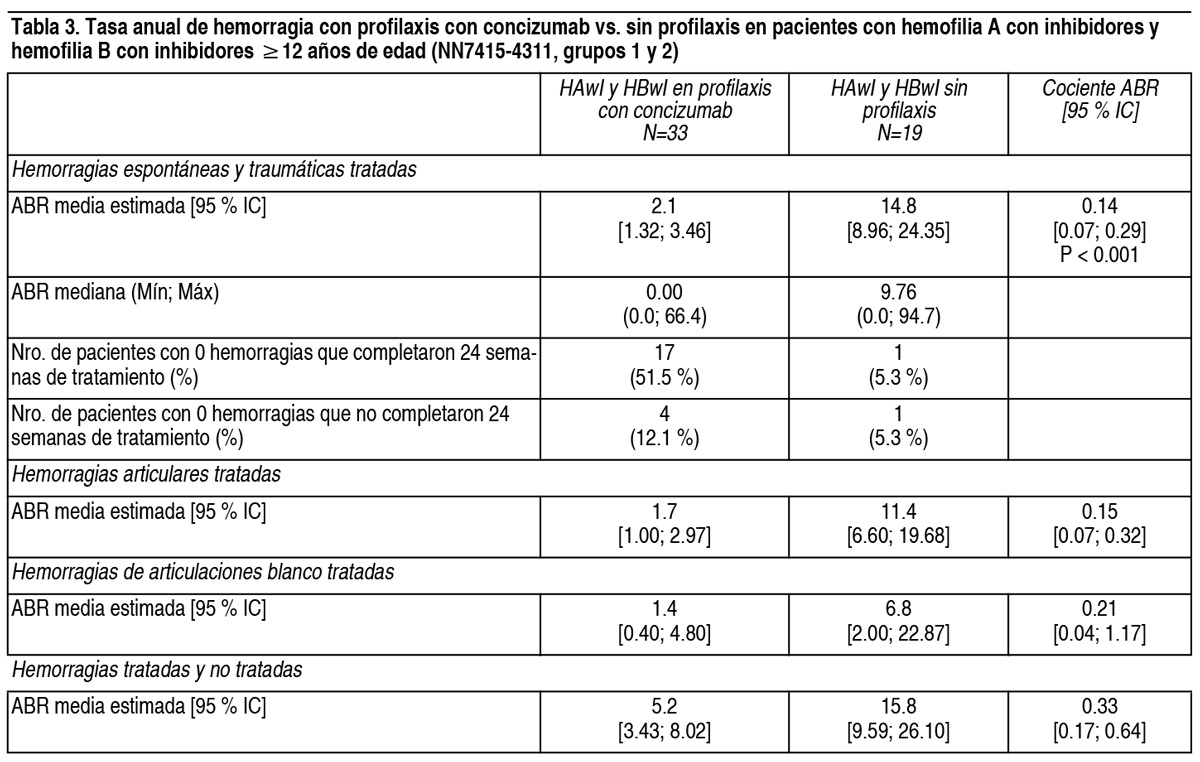
Propiedades:Propiedades farmacodinámicas: Mecanismo de acción: Concizumab es un anticuerpo dirigido contra el inhibidor de la vía del factor tisular (anti-TFPI). El TFPI es un inhibidor del factor Xa (FXa). La unión de concizumab al TFPI evita la inhibición del FXa por parte del TFPI. El aumento de la actividad del FXa prolonga la fase de iniciación de la coagulación y permite la generación de suficiente trombina para una hemostasia efectiva. Concizumab actúa independientemente del FVIII y el FIX. Datos farmacodinámicos: En el estudio NN7415-4311, la media de TFPI libre (TFPI plasmático no unido a concizumab) para los pacientes que recibieron profilaxis con concizumab disminuyó un 87 % dentro de las 24 horas posteriores a la administración de la dosis de carga de concizumab y se mantuvo estable a lo largo del tiempo. Concizumab restableció la capacidad de generación de trombina, reflejada por un pico medio de trombina dentro del rango del plasma normal y el 94 % de los pacientes presentando valores pico de trombina dentro del rango del plasma normal (26-147 nM) en el punto de corte a las 56 semanas. Transitoriamente, se informaron niveles pico de trombina moderadamente elevados en el 37.6 % de los pacientes, sin riesgos de seguridad asociados. Datos de eficacia clínica y seguridad: Pacientes con hemofilia A y B con inhibidores (HAwI y HBwI) de 12 ańos o más (NN74515-4311): El estudio NN7415-4311 fue un estudio de fase 3 multinacional, multicéntrico, abierto, para investigar la eficacia y la seguridad de concizumab para la profilaxis de episodios hemorrágicos en 91 pacientes adultos (58 con HAwI y 33 con HBwI) y 42 adolescentes (22 con HAwI y 20 con HBwI) masculinos con hemofilia A o B con inhibidores. El estudio estaba compuesto por 4 grupos, incluyendo 2 grupos no aleatorizados: Los grupos 1 y 2: 52 pacientes tratados previamente a demanda, fueron aleatorizados a sin profilaxis (tratamiento a demanda con agentes bypass; grupo 1) o profilaxis con concizumab (grupo 2), con ≥6 hemorragias tratadas en las últimas 24 semanas o ≥12 hemorragias tratadas en las últimas 52 semanas antes del reclutamiento, o que fueron transferidos del estudio NN7415-4322. Grupos 3 y 4: 81 pacientes adicionales (53 con HAwI y 28 con HBwI) tratados con profilaxis con concizumab. Los pacientes tenían 12 ańos de edad o más y un peso corporal >25 kg, con hemofilia A o B congénita de cualquier severidad con antecedentes documentados de inhibidores (≥0.6 BU), que habían sido prescritos o necesitaron tratamiento con agentes de bypass en las últimas 24 semanas anteriores al reclutamiento. Los pacientes recibieron un regimen posológico de acuerdo con las recomendaciones del prospecto. El objetivo primario del estudio fue comparar el efecto de la profilaxis con concizumab con la ausencia de profilaxis (tratamiento a demanda con agentes de bypass) respecto de la reducción del número de episodios de sangrado en pacientes adultos y adolescentes con hemofilia A y B con inhibidores (ver Tabla 3). Mediante un modelo binomial negativo, se estimó un cociente de las tasas anuales de hemorragia (ABR) de 0.14 (p <0.001), lo que corresponde a una reducción de la ABR del 86 % para los sujetos que recibieron profilaxis con concizumab en comparación con la ausencia de profilaxis. Un análisis adicional incluyendo toda la información disponible siguiendo el principio ITT (intención de tratar) muestra un cociente de la ABR estimado de 0.20 (95 % IC [0.09;0.45], p<0.001). Adicionalmente, se calculó el número de pacientes con cero hemorragias. La mediana de ABR y la cantidad de pacientes con cero hemorragias se muestran en la Tabla 3. La eficacia también se evaluó cuando todos los pacientes de los grupos 2, 3 y 4 habían completado al menos 56 semanas de tratamiento, y los resultados fueron consistentes con los resultados presentados en la Tabla 3.
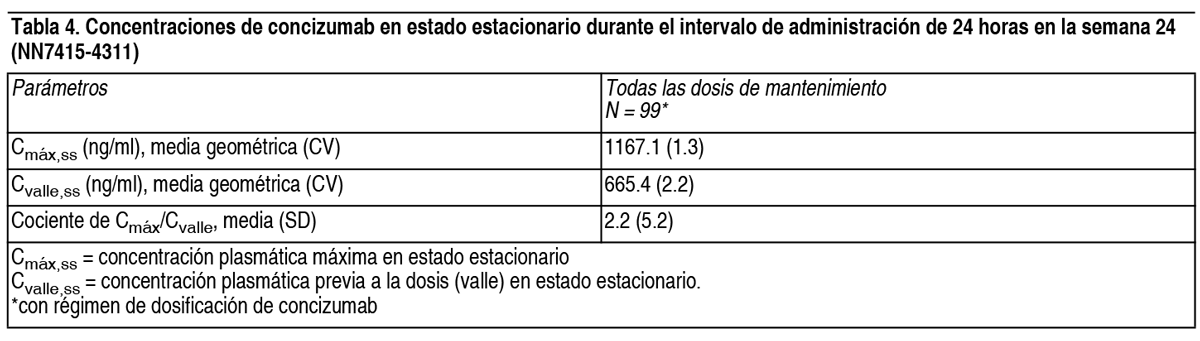
Valores de laboratorio de dímero D de fibrina y fragmentos 1+2 de protrombina aumentados: Se informaron niveles aumentados de dímero D de fibrina y fragmentos 1+2 en los estudios de dosis múltiples. La concentración plasmática de concizumab se correlaciona positivamente con el dímero D de fibrina y los fragmentos 1+2 de protrombina, lo que indica un efecto hemostático de concizumab. No se observaron cambios clínicamente significativos en el fibrinógeno, la antitrombina y las plaquetas. Población pediátrica: La seguridad y la eficacia en nińos menores de 12 ańos aún no se han estudiado en un estudio clínico. Tratamiento de hemorragias intercurrentes en estudios clínicos: Mientras se utilizaba el régimen de dosificación de concizumab y la guía de hemorragias intercurrentes de la sección Posología y modo de administración, las hemorragias se trataron de manera efectiva y segura, y no se observaron eventos tromboembólicos. La seguridad y eficacia del uso concomitante del régimen posológico de profilaxis con concizumab y el tratamiento de hemorragias intercurrentes se confirmaron en el estudio NN7415-4311. Un total de 408 episodios hemorrágicos se trataron con rFVIIa (mayoría) y FEIBA (≥56 semanas para los grupos de tratamiento con concizumab). Inmunogenicidad: Durante los períodos de tratamiento en los estudios NN7415-4159 (11 semanas), NN7415-4310 y NN7415-4255 (≥76 semanas), NN7415-4311 (≥56 semanas para los grupos de tratamiento con concizumab) y NN7415-4307 (≥32 semanas para los grupos de tratamiento con concizumab), 68 de los 320 pacientes tratados con concizumab (21.3 %) resultaron positivos para anticuerpos anti-concizumab, de los cuales 17 pacientes (5.3 %) resultaron positivos para anticuerpos neutralizantes in vitro. En 1 (1.5 %) de los 68 pacientes positivos para anticuerpos anti-concizumab, los anticuerpos neutralizantes in vitro coocurrieron con la restauración de los niveles de TFPI libre. En los 67 pacientes restantes (98.5 %), no se identificó ningún efecto clínicamente significativo de los anticuerpos sobre la farmacocinética, farmacodinámica, seguridad o efectividad de concizumab. Propiedades farmacocinéticas: Los estudios farmacocinéticos han demostrado que la exposición sistémica a concizumab, medida por el AUC y la Cmáx, aumentó más que proporcionalmente a la dosis. Este comportamiento farmacocinético no lineal es causado por la disposición del fármaco mediada por el blanco (TMDD), que ocurre cuando concizumab se une al TFPI unido a células endoteliales con posterior eliminación del complejo fármaco-blanco. Este es un proceso saturable y el alcance de la eliminación de concizumab mediante TMDD se determina por la cantidad de TFPI unido a células endoteliales. Esto da lugar a una eliminación rápida/alto clearance a concentraciones bajas de concizumab (donde la vía no lineal es dominante) y una eliminación más lenta/menor clearance a concentraciones más altas de concizumab (donde la vía lineal es dominante). La exposición a concizumab fue similar entre la hemofilia A y la B con inhibidores. Las concentraciones medias geométricas de concizumab en estado estacionario, en la semana 24, se muestran en la Tabla 4. Las concentraciones plasmáticas previas a la dosis (valle) se mantuvieron estables durante las 56 semanas de tratamiento.
Absorción: Tras la administración s.c. de una dosis única de 0.05-3 mg/kg de concizumab en sujetos sanos y con hemofilia, el tiempo hasta la concentración plasmática máxima de concizumab (tmáx) estuvo en el rango de 8 horas a 99 horas (4.1 días). Metabolismo: Concizumab es un anticuerpo y, al igual que otras proteínas grandes, estas se catabolizan principalmente mediante proteólisis lisosomal a aminoácidos, que posteriormente el cuerpo excreta o reutiliza. Se espera que concizumab siga esta vía catabólica tanto para la vía de eliminación no lineal a través de TMDD como para la vía de eliminación lineal a través de la unión al receptor Fc, que es frecuente para los anticuerpos. Eliminación: Tanto la vía lineal como la no lineal contribuyen a la eliminación de concizumab. La vida media terminal en sujetos sanos y con hemofilia que recibieron una dosis s.c. única de 0.25-3 mg/kg se midió en el rango de 39 horas (1.6 días) a 195 horas (8.1 días). En los niveles en estado estacionario, donde la eliminación lineal se vuelve dominante, la vida media total puede ser más prolongada. Poblaciones especiales: Edad: La edad no tuvo ningún efecto sobre la exposición a concizumab en pacientes con hemofilia A o B con inhibidores. La población del estudio se encontraba en el rango etario de 12 a 61 ańos. Insuficiencia renal: Los datos disponibles en insuficiencia renal son limitados. De los 112 pacientes tratados con el régimen de dosificación de concizumab en NN7415-4311, 4 pacientes tenían insuficiencia renal leve (TFGe entre 60 y 90 ml/min/1.73 m2) y 1 paciente tenía insuficiencia renal moderada (TFGe entre 30 y 60 ml/min/1.73 m2) al momento de la administración de la dosis de carga. No se observó ningún impacto en la exposición a concizumab. No hay datos disponibles en insuficiencia renal grave. Insuficiencia hepática: Los datos en insuficiencia hepática son limitados o inexistentes. De los 112 pacientes tratados con el régimen de dosificación de concizumab en NN7415-4311, 4 pacientes presentaron enzimas hepáticas aumentadas (ALT o AST ≥1.5 x ULN) al momento de la administración de la dosis de carga. No se observó ningún impacto en la exposición a concizumab. Datos preclínicos de seguridad: Los datos preclínicos no revelan ningún riesgo especial para los seres humanos, según los estudios convencionales de toxicología a dosis repetidas. Se observó formación de trombos mediada farmacológicamente en un estudio de toxicología de 52 semanas en monos cynomolgus a dosis subcutáneas ≥1 mg/kg/día (correspondiente a 300 veces la exposición humana basada en el AUC0-24h). Carcinogenicidad: No se han realizado estudios en animales para evaluar el potencial carcinogénico de concizumab, ni estudios para determinar los efectos de concizumab sobre la genotoxicidad. Fertilidad: En un estudio de toxicidad de 26 semanas en monos cynomolgus sexualmente maduros, machos y hembras, con dosis subcutáneas de hasta 9 mg/kg/día (correspondientes a 3400 veces la exposición humana, basada en el AUC0-24h), concizumab no afectó la fertilidad (tamańo testicular, funcionalidad del esperma o duración del ciclo menstrual) y no causó ningún cambio en los órganos reproductivos masculinos ni femeninos. Teratogenicidad: No hay datos disponibles con respecto a los posibles efectos secundarios de concizumab en el desarrollo embriofetal. Interacciones medicamentosas: En un estudio de toxicidad de interacción medicamentosa de 28 días en monos cynomolgus con administración diaria de 1 mg/kg de concizumab para alcanzar el estado estacionario, se administraron 3 dosis intravenosas consecutivas de hasta 1 mg/kg de rFVIIa, a intervalos de 2 horas, a los animales tratados con concizumab. No se observaron hallazgos adversos con una exposición a concizumab correspondiente a 200 veces la exposición humana, basada en el AUC0-24h.
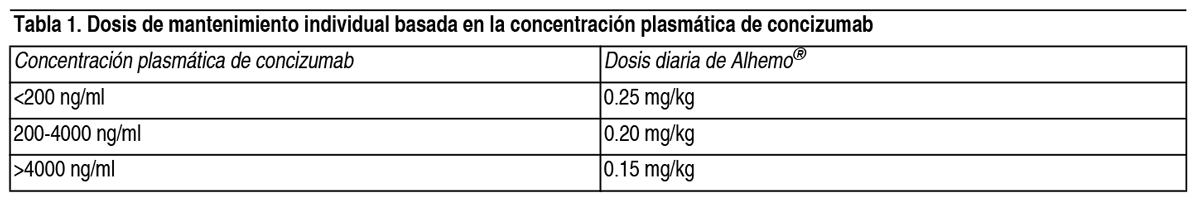
Posología: El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia y/o trastornos hemorrágicos. El tratamiento debe iniciarse en un estado no hemorrágico. El tratamiento con rFVIIa debe interrumpirse al menos 12 horas antes de iniciar la terapia con concizumab, y el tratamiento con aPCC debe interrumpirse al menos 48 horas antes. El régimen de dosificación recomendado es: Día 1: una dosis de carga de 1 mg/kg 1 vez. Día 2 y hasta el ajuste de la dosis de mantenimiento individual (ver abajo): 1 dosis por día de 0.20 mg/kg. 4 semanas después del inicio del tratamiento: Medición de la concentración plasmática de concizumab antes de la administración de la siguiente dosis programada. La medición debe realizarse utilizando un test diagnóstico in vitro validado. Cuando el resultado de la concentración plasmática de concizumab esté disponible: La dosis de mantenimiento individual se establece una vez en función de la concentración plasmática de concizumab, según se indica a continuación en la Tabla 1.
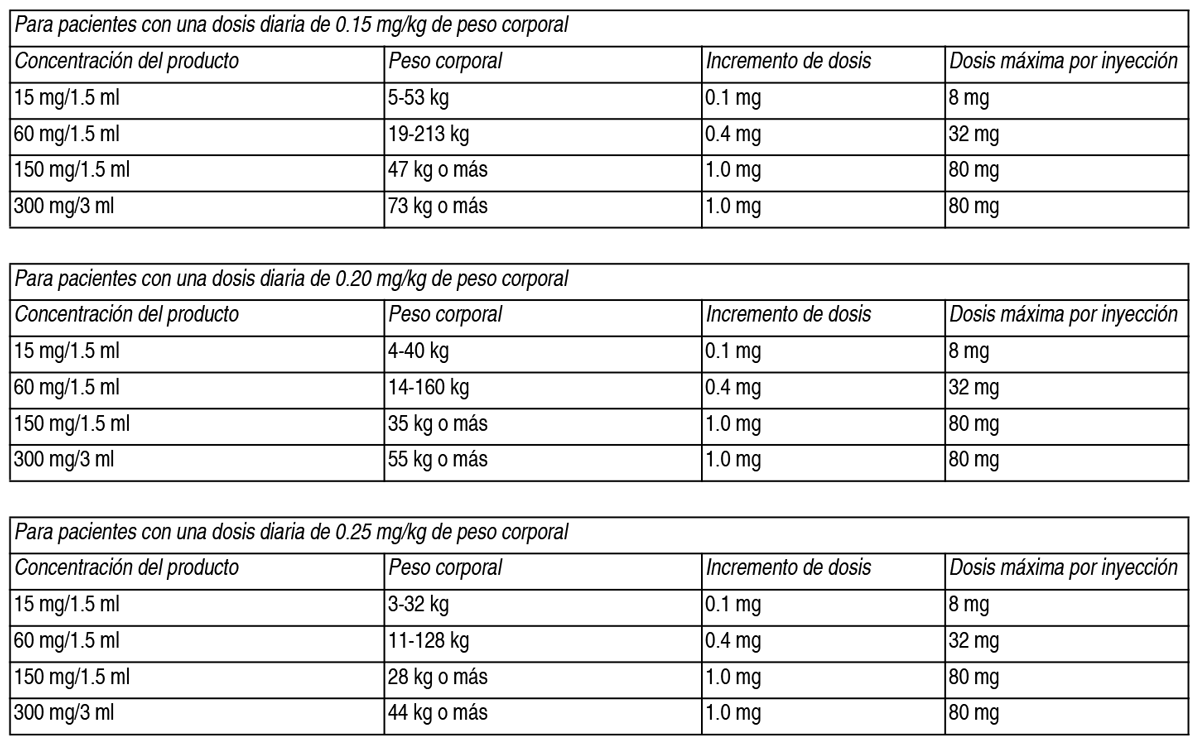
El ajuste de la dosis de mantenimiento individual debe realizarse lo antes posible (después de que esté disponible el resultado de la concentración plasmática de concizumab), y se recomienda que sea no más de 8 semanas después del inicio del tratamiento. Pueden realizarse determinaciones adicionales de la concentración plasmática de concizumab después de 8 semanas con la misma dosis de mantenimiento según la condición médica del paciente. Por ejemplo, esto puede considerarse si un paciente experimenta un aumento en la frecuencia de hemorragia, un gran cambio en el peso corporal, ha omitido dosis antes de establecer la dosis de mantenimiento o adquiere una comorbilidad que puede conducir a un aumento en el riesgo tromboembólico total. Dado que concizumab es dosificado en función del peso corporal (mg/kg), es importante recalcular la dosis (mg) cuando el peso corporal cambia. Cálculo de la dosis: La dosis (en mg) se calcula de la siguiente manera: Peso corporal del paciente (kg) x dosis (1, 0.15, 0.20 o 0.25 mg/kg) = cantidad total (mg) de concizumab a administrar. La dosis se selecciona en incrementos de: 0.1 mg en la lapicera de 15 mg/1.5 ml (color azul), 0.4 mg en la lapicera de 60 mg/1.5 ml (color marrón) y 1.0 mg en las lapiceras de 150 mg/1.5 ml y 300 mg/3 ml (color dorado). La dosis calculada se redondea a la dosis inyectable de la lapicera más cercana. El médico o enfermero debe ayudar al paciente a redondear e identificar la dosis inyectable adecuada en la lapicera. Idealmente, los pacientes deberían recibir la prescripción y utilizar una lapicera que pueda administrar la dosis de mantenimiento diaria requerida en una sola inyección. La dosis inyectable más cercana puede identificarse girando el tambor de la lapicera o calcularse como se indica a continuación: Divida la dosis total en mg por la dosis del incremento. Redondee al número entero más cercano. Multiplique por la dosis del incremento. Ejemplos: Paciente con un peso corporal de 42 kg, utilizando una dosis de mantenimiento de 0.15 mg/kg. Día 1, utilizando una dosis de carga de 1 mg/kg: 42 kg x 1 mg/kg = 42 mg de concizumab. Día 2 y hasta que se establezca la dosis de mantenimiento individual, utilizando una dosis de 0.20 mg/kg: 42 kg x 0.20 mg/kg = 8.4 mg de concizumab. Dosis de mantenimiento: 42 kg x 0.15 mg/kg = 6.3 mg de concizumab. El paciente debe recibir 6.3 mg de concizumab con una lapicera de 60 mg/1.5 ml para obtener la mayor duración de la lapicera (en días) para el peso corporal de este paciente. Para identificar la dosis inyectable más cercana: 6.3 mg dividido 0.4 mg/incremento = 15.75 incrementos. 15.75 incrementos se redondea a 16 incrementos. 16 incrementos multiplicado por 0.4 mg/incremento = 6.4 mg. 6.4 mg es una dosis seleccionable en la lapicera de 60 mg/1.5 ml y es la dosis inyectable más cercana a 6.3 mg. Paciente con un peso corporal de 67 kg, utilizando una dosis de mantenimiento de 0.20 mg/kg: Día 1, utilizando una dosis de carga de 1 mg/kg: 67 kg x 1 mg/kg = 67 mg de concizumab. Día 2 y hasta que se establezca la dosis de mantenimiento individual, utilizando una dosis de 0.20 mg/kg: 67 kg x 0.20 mg/kg = 13.4 mg de concizumab. Dosis de mantenimiento: 67 kg x 0.20 mg/kg = 13.4 mg de concizumab. El paciente debe recibir 13.4 mg de concizumab con una lapicera de 300 mg/3 ml para obtener la mayor duración de la lapicera (en días) para el peso corporal de este paciente. Para identificar la dosis inyectable más cercana: 13.4 mg dividido 1.0 mg/incremento = 13.4 incrementos-13.4 incrementos se redondea a 13 incrementos. 13 incrementos multiplicado por 1.0 mg/incremento = 13.0 mg. 13.0 mg es una dosis seleccionable en la lapicera de 300 mg/3 ml y es la dosis inyectable más cercana a 13.4 mg. Elección de la concentración y el volumen del producto: En función de las características técnicas, las lapiceras Alhemo® pueden adaptarse a los siguientes rangos de peso corporal:
Si más de 1 lapicera Alhemo® es relevante con base en el rango de peso corporal, se debería seleccionar la lapicera con la mayor concentración del producto. La lapicera con mayor concentración contiene más dosis que pueden administrarse, lo que permite que la lapicera se use por más días. Duración del tratamiento: Alhemo® está propuesto para el tratamiento profiláctico a largo plazo. Dosis omitida: Concizumab se puede administrar en cualquier momento del día. Es importante la adherencia de cada paciente a su dosificación diaria. La adherencia es particularmente importante durante las 4 semanas iniciales para asegurar que se establezca una dosis de mantenimiento correcta con base en la concentración plasmática de concizumab en la semana 4 (ver Posología más arriba). Los pacientes que omitan dosis antes del establecimiento de la dosis de mantenimiento deben reanudar el tratamiento lo antes posible con la dosis diaria inicial de 0.2 mg/kg e informar a su profesional de la salud. Dosis omitidas luego de establecida la dosis de mantenimiento: La siguiente guía de dosificación solo aplica cuando un paciente ha olvidado u omitido la administración de su dosis diaria de mantenimiento. 1 dosis diaria omitida: El paciente debe retomar la dosis diaria de mantenimiento sin una dosis adicional. 2 a 6 dosis diarias consecutivas omitidas: El paciente debe administrarse 2 veces la dosis diaria (como 2 inyecciones separadas, cada una correspondiendo a 1 dosis diaria) y luego continuar administrándose la dosis de mantenimiento diaria al día siguiente. 7 o más dosis diarias consecutivas omitidas: El paciente debe contactar a su profesional de la salud inmediatamente. El paciente puede necesitar recibir una nueva dosis de carga antes de continuar con su dosis de mantenimiento diaria al día siguiente, luego de una cuidadosa consideración del cuadro clínico. En caso de duda, el paciente debe contactar a su profesional de la salud. Manejo de hemorragias intercurrentes: No se debe realizar ningún ajuste de la dosis de Alhemo® en caso de hemorragias intercurrentes. Los médicos deben conversar con el paciente y/o el cuidador sobre la dosis y el cronograma de los agentes bypass, si es necesario, mientras reciben profilaxis con concizumab. El tratamiento con agentes bypass (p. ej., rFVIIa o aPCC) puede usarse para hemorragias intercurrentes, y la dosis y la duración dependerán de la ubicación y la gravedad de la hemorragia. Para las hemorragias leves y moderadas que requieren tratamiento adicional con agentes bypass (p. ej., rFVIIa o aPCC), se recomienda utilizar la dosis más baja y el intervalo de dosificación indicados en el prospecto aprobado. Además, para el aPCC, se recomienda una dosis máxima de 100 U/kg de peso corporal en un plazo de 24 horas. Para las hemorragias graves, se recomienda seguir el esquema de dosificación proporcionado en el prospecto aprobado para el producto específico, en función del criterio clínico. Manejo en el entorno perioperatorio: No es necesario ajustar la dosis de Alhemo® en caso de cirugías menores. Antes de una cirugía mayor, debe consultarse a un profesional de la salud con experiencia en el tratamiento de la hemofilia y/o trastornos hemorrágicos. Dado que la experiencia clínica utilizando concizumab durante cirugías mayores es limitada, generalmente se recomienda pausar la administración de concizumab al menos 4 días antes de una cirugía mayor programada. El tratamiento con concizumab puede reanudarse 10-14 días después de la cirugía con la misma dosis de mantenimiento, sin una nueva dosis de carga, considerando el cuadro clínico general del paciente. Los criterios que definen una cirugía mayor son cualquier procedimiento quirúrgico invasivo que requiera ≥3 dosis de terapia bypass y/o cuando ocurre 1 o más de los siguientes: Se accede a una cavidad corporal. Se cruza una barrera mesenquimática (por ejemplo, pleura, peritoneo o duramadre). Se abre un plano de fascia. Se extirpa un órgano. Se altera quirúrgicamente la anatomía normal. Inducción de tolerancia inmunitaria (ITI): No se han establecido la seguridad y la eficacia del uso concomitante de concizumab en pacientes que se encuentren recibiendo ITI, una estrategia de desensibilización para la erradicación de inhibidores. No hay datos disponibles. Se deben evaluar cuidadosamente los potenciales beneficios y riesgos si se considera la continuación o iniciación de concizumab durante la ITI. Poblaciones especiales: Pacientes pediátricos: Aún no se ha establecido la eficacia y la seguridad de Alhemo® en nińos <12 ańos con hemofilia A con inhibidores y hemofilia B con inhibidores. No hay datos disponibles. Pacientes de edad avanzada: No se recomiendan ajustes de dosis (además del ajuste de dosis de mantenimiento individual) en pacientes ≥65 ańos. No hay datos disponibles en pacientes de 65 ańos o más. Para más información, ver sección Propiedades farmacocinéticas. Insuficiencia renal: No se recomiendan ajustes de dosis (además del ajuste de dosis de mantenimiento individual) en pacientes con insuficiencia renal. Los datos disponibles en pacientes con insuficiencia renal leve, moderada y severa son limitados o inexistentes; consulte la sección Propiedades farmacocinéticas. Insuficiencia hepática: No se recomiendan ajustes de dosis (además del ajuste de dosis de mantenimiento individual) en pacientes con insuficiencia hepática. Los datos disponibles en pacientes con insuficiencia hepática son limitados o inexistentes; consulte la sección Propiedades farmacocinéticas. Modo de administración: Alhemo® es para uso subcutáneo únicamente. El concizumab viene en una lapicera prellenada lista para administrar. Las agujas no están incluidas; consulte la sección Presentación. Concizumab debe administrarse diariamente, en cualquier momento del día, no necesariamente a la misma hora todos los días. Concizumab puede ser autoadministrado, o administrado por un cuidador, después de recibir el entrenamiento adecuado por parte de un profesional de la salud y de leer las instrucciones de uso. Concizumab debe administrarse mediante inyección subcutánea en el abdomen o el muslo con rotación del sitio de inyección todos los días. Las inyecciones subcutáneas no deben administrarse en zonas donde la piel esté sensible, con hematomas, enrojecida o dura, ni en zonas donde haya lunares o cicatrices. Siempre debe utilizarse una aguja nueva para cada inyección. Cada lapicera de Alhemo® debe ser utilizada por un solo paciente. Las lapiceras de Alhemo® no deben compartirse entre pacientes, incluso si se cambia la aguja. Para obtener instrucciones detalladas sobre la administración del medicamento, consulte la sección Precauciones especiales de descarte y manipulación y el prospecto incluido en el envase.
Modo de Empleo:Precauciones especiales de descarte y manipulación: Para una inyección más confortable, permitir que el medicamento tome temperatura ambiente si estaba almacenado en la heladera. No utilice fuentes de calor artificiales. Inspeccionar la solución visualmente antes del uso. A través de la ventana de la lapicera, Alhemo® se presenta como una solución transparente a ligeramente opalescente e incolora a ligeramente amarilla y prácticamente libre de partículas visibles. Las partículas de proteína translucidas a blancas son aceptables. No utilizar si el medicamento presenta alteraciones en el color. En las Instrucciones de uso, se proporcionan instrucciones detalladas para la preparación y administración del medicamento. Se debe indicar a los pacientes delgados y a los adolescentes que utilicen técnicas de inyección que minimicen el riesgo de inyección intramuscular; por ejemplo, inyectando en un pliegue de piel suelto. Todo medicamento no utilizado o los materiales de desecho se deben descartar de acuerdo con los requisitos locales.
Efectos Colaterales:Resumen del perfil de seguridad: El perfil de seguridad general de concizumab se basa en los datos de los estudios clínicos. Las reacciones adversas más graves en los estudios clínicos con concizumab fueron eventos tromboembólicos (0.9 %) e hipersensibilidad (0.3 %). Resumen tabulado de reacciones adversas: Las siguientes reacciones adversas se basan en datos agrupados de los estudios clínicos NN7415-4159 (fase 1b), NN7415-4310 (fase 2), NN7415-4255 (fase 2), NN7415-4311 (fase 3) y NN7415-4307 (fase 3), en los que un total de 320 pacientes masculinos con hemofilia A con y sin inhibidores y hemofilia B con y sin inhibidores recibieron al menos 1 dosis de concizumab como profilaxis de rutina. Los pacientes estuvieron expuestos por un total de 411 ańos de exposición. La siguiente tabla se presenta según la clasificación de órganos y sistemas de MedDRA (SOC y Nivel de Término Preferente). Las frecuencias se han evaluado de acuerdo con la siguiente convención: Muy frecuentes (≥1/10), frecuentes (≥1/100 a <1/10), poco frecuentes (≥1/1000 a <1/100), raras (≥1/10 000 a <1/1000), muy raras (<1/10 000) y frecuencia desconocida (no puede estimarse a partir de los datos disponibles). Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad; vea la Tabla 2.
Descripción de reacciones adversas seleccionadas: Reacciones en el sitio inyección: Se informaron reacciones en el sitio de inyección en los estudios clínicos de dosis múltiples. Los síntomas notificados con mayor frecuencia fueron eritema en el sitio de inyección (5.9 %), moretones en el sitio de inyección (4.4 %) y hematomas en el sitio de inyección (4.1 %). La mayoría se informaron como leves. Población pediátrica: 78 de los participantes en estudios clínicos eran adolescentes (≥12 a <18 ańos). El perfil de seguridad fue similar entre los pacientes adolescentes y adultos, y fue el esperado para el grupo etario. La seguridad y eficacia de concizumab en nińos menores de 12 ańos no fue establecida aún. No hay datos disponibles. Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas después de la autorización del medicamento. Esto permite un monitoreo continuo del balance riesgo/beneficio del medicamento. Se solicita a los profesionales de la salud que notifiquen cualquier sospecha de reacción adversa.
Contraindicaciones: Hipersensibilidad al principio activo o a cualquiera de los excipientes listados en la sección Composición.
Advertencias:Reacciones de hipersensibilidad: Se han producido reacciones de hipersensibilidad de tipo alérgico con concizumab durante las semanas iniciales de tratamiento, incluyendo hospitalización y discontinuación permanente del tratamiento. Se debe informar a los pacientes sobre los signos de reacciones de hipersensibilidad aguda. Si se producen síntomas de hipersensibilidad, se debe indicar al paciente que interrumpa el uso de Alhemo® y que se comunique con el médico, quien debe garantizar el tratamiento adecuado. Inmunogenicidad: El desarrollo de anticuerpos neutralizantes anti-concizumab observado en algunos pacientes no ha conducido a pérdida de eficacia (ver sección Propiedades farmacodinámicas). Sin embargo, los pacientes con signos clínicos de pérdida de eficacia (por ejemplo, aumento de los eventos de hemorragia intercurrente) deben ser evaluados para determinar la etiología, y deberían considerarse otras opciones terapéuticas si se sospecha la presencia de anticuerpos neutralizantes anti-concizumab. Eventos tromboembólicos: Se han informado eventos tromboembólicos arteriales y venosos no mortales en los estudios clínicos de concizumab. Estos casos ocurrieron en pacientes con múltiples factores de riesgo, incluyendo el uso de dosis altas o frecuentes de tratamiento de hemorragias intercurrentes (ver sección Reacciones adversas). Se debe informar y controlar a los pacientes tratados con concizumab sobre la aparición de signos y síntomas de eventos tromboembólicos. En caso de sospecha de eventos tromboembólicos, se debe interrumpir el uso de concizumab y se deben iniciar más investigaciones y el tratamiento médico adecuado. Debe considerarse cuidadosamente si el potencial beneficio del tratamiento con concizumab supera el potencial riesgo en pacientes considerados de alto riesgo para eventos tromboembólicos. Esta consideración debe reevaluarse periódicamente. En condiciones en las que el factor tisular está sobreexpresado (p. ej., enfermedad ateroesclerótica avanzada, lesión por aplastamiento, cáncer o septicemia), puede existir un riesgo de eventos tromboembólicos o coagulación intravascular diseminada (CID). En estas situaciones, debe contrastarse el potencial beneficio del tratamiento con concizumab contra el riesgo de estas complicaciones. Contenido de sodio: Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis; es decir, es esencialmente «libre de sodio».
Precauciones:Embarazo, lactancia y fertilidad: Mujeres en edad fértil/anticoncepción en hombres y mujeres: Las mujeres en edad fértil que reciben concizumab deben utilizar métodos anticonceptivos altamente efectivos durante el tratamiento con concizumab y hasta 7 semanas después del fin del tratamiento. El médico tratante debe evaluar los riesgos tromboembólicos y beneficios del tipo de anticonceptivos utilizado. Embarazo: No hay datos disponibles sobre el uso de concizumab en mujeres embarazadas. No se han realizado estudios de reproducción animal con concizumab. Se desconoce si concizumab puede causar dańo fetal cuando se administra a una mujer embarazada o si puede afectar la capacidad reproductiva. Concizumab solo debería utilizarse durante el embarazo si el beneficio potencial para la madre supera el riesgo potencial para el feto. Lactancia: Se desconoce si concizumab se excreta en la leche materna. Se conoce que las IgG humanas se excretan en la leche materna durante los primeros días después del parto, decreciendo a concentraciones bajas poco después; consecuentemente, no puede excluirse un riesgo para el lactante durante este corto período. Posteriormente, concizumab podría utilizarse durante la lactancia si es clínicamente necesario. Fertilidad: Los estudios en animales no indican efectos perjudiciales directos o indirectos con respecto a la fertilidad; consulte la sección Datos preclínicos de seguridad. No hay datos disponibles de fertilidad en seres humanos. Por lo tanto, se desconoce el efecto de concizumab sobre la fertilidad masculina y femenina. Efectos sobre la capacidad para conducir y utilizar máquinas: Alhemo® no tiene influencia alguna en la capacidad para conducir vehículos y utilizar máquinas.
Interacciones Medicamentosas: No se han realizado estudios clínicos de interacción farmacológica. Se realizó un estudio de toxicidad de interacción farmacológica con rFVIIa en monos cynomolgus tratados con concizumab. No se observaron signos de trombosis ni otros hallazgos adversos en monos normocoagulantes al agregar 3 dosis consecutivas de hasta 1 mg/kg de rFVIIa sobre concizumab en estado estacionario; consulte la sección Datos preclínicos de seguridad. Se realizaron estudios de interacción farmacológica in vitro y ex vivo con rFVIIa, aPCC, rFVIII o rFIX en sangre de pacientes con hemofilia que recibían tratamiento profiláctico con concizumab. Estos estudios no indicaron interacciones farmacológicas clínicamente relevantes. Para obtener orientación sobre el uso de agentes bypass para el tratamiento de episodios hemorrágicos intercurrentes en pacientes que reciben profilaxis con concizumab, consulte la sección Posología y modo de administración.
Sobredosificación: La experiencia con sobredosis de concizumab es limitada. Se han informado casos de hasta 5 veces la dosis prevista sin consecuencias clínicas. La sobredosis accidental podría resultar en hipercoagulabilidad y los pacientes deben contactar a su médico para ser monitoreados. Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con un centro de toxicología: Hospital de Pediatría «Ricardo Gutiérrez»: (011) 4962 6666 / 2247. Hospital «A. Posadas»: (011) 4654 6648 / 4658 7777.
Incompatibilidades: En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros.
Conservación:Antes del primer uso: Conservar en heladera (entre 2 °C y 8 °C). Después del primer uso: Conservar por hasta 4 semanas a una temperatura menor a 30 °C o en heladera (entre 2 °C y 8 °C). Se ha demostrado la estabilidad química y física en uso por 28 días a 30 °C o en heladera. Desde un punto de vista microbiológico, una vez abierto, el producto puede ser almacenado por un tiempo máximo de 28 días a 30 °C o en heladera. Otros tiempos y condiciones de almacenamiento en uso son responsabilidad del usuario. Conservar la lapicera con el capuchón colocado para proteger la solución de la luz. Conservar la lapicera sin una aguja colocada. Esto garantiza la dosificación exacta y previene la contaminación, las infecciones y la pérdida de producto. No congelar. Mantener alejado del elemento refrigerante de la heladera. Alhemo® debe protegerse del calor y la luz y no debe exponerse a la luz solar directa.
Observaciones: Mantener fuera del alcance y la vista de los nińos.
Presentaciones: Alhemo® se presenta en una lapicera prellenada descartable multidosis conteniendo un cartucho de vidrio de 1.5 ml o 3 ml. La lapicera prellenada se proporciona en un envase. Alhemo® está disponible en las siguientes presentaciones, y el botón pulsador y el soporte del cartucho de la lapicera están codificados por colores según la concentración: 15 mg/1.5 ml (azul): 1 lapicera por envase. 60 mg/1.5 ml (marrón): 1 lapicera por envase. 150 mg/1.5 ml (dorada): 1 lapicera por envase. 300 mg/3 ml (dorada): 1 lapicera por envase. Es posible que no todas las presentaciones se encuentren comercializadas. Las agujas para inyección no están incluidas. Se recomienda usar Alhemo® con agujas NovoFine® Plus o NovoFine® de calibre 32 y una longitud de 4 mm. Si se usan agujas de más de 4 mm de longitud, se deben utilizar técnicas de inyección que minimicen el riesgo de inyección intramuscular; por ejemplo, inyectando en un pliegue de piel suelto.
 Laboratorio
Laboratorio