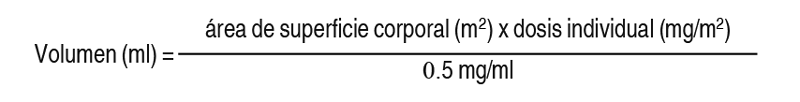Oncológicos y Terapias Relacionadas: Citostáticos Antineoplásicos
Información farmacológica
Composición: Cada vial de dosis única contiene: Lurbinectedina 4 mg. Excipientes: Sacarosa 800 mg, Ácido Láctico 22.1 mg e Hidróxido de Sodio 5.1 mg.
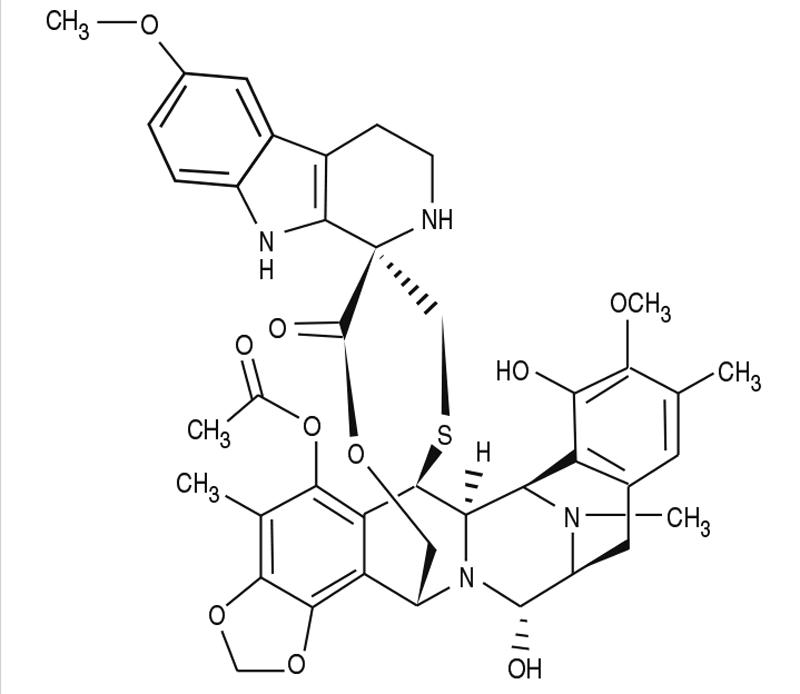
Descripción: Zepzelca es un fármaco alquilante. El nombre químico de Zepzelca (lurbinectedina) es (1'R,6R,6aR,7R,13S,14S,16R)-8,14-dihidroxi-6',9-dime- toxi-4,10,23-trimetil-19-oxo2',3',4',6,7,9',12,13,14,16-decahidro-6aH-espiro [7,13-azano-6,16-(epitiopropanoximetano) [1,3]dioxolo[7,8]isoquinoleína[3,2-b][3]benzocaína-20,1'-pirido[3,4-b]indol]-5-yl acetato. La fórmula molecular es C41H44N4O10S. El peso molecular es de 784.87 g/mol y la estructura química es la siguiente:
Zepzelca de 4 mg para inyección se suministra como un polvo liofilizado en un vial de dosis única para la reconstitución y uso intravenoso. La formulación liofilizada de Zepzelca está compuesta de lurbinectedina de 4 mg, sacarosa (800 mg), ácido láctico (22.1 mg) e hidróxido de sodio (5.1 mg). Antes de su uso, la liofilización se reconstituye al agregar 8 ml de agua para inyección USP, lo que forma una solución que contiene 0.5 mg/ml de lurbinectedina (la concentración calculada es de 0.47 mg/ml en función del volumen final de 8.5 ml).
Indicaciones: Zepzelca está indicado para el tratamiento de pacientes adultos con cáncer de pulmón de células pequeńas (SCLC) metastásico, con progresión de la enfermedad durante o después del tratamiento de quimioterapia con un derivado del platino.
Propiedades:Mecanismo de acción: La lurbinectedina es un fármaco alquilante que se une a los residuos de guanina en el surco menor del ADN, lo que forma aductos y provoca una flexión de la hélice del ADN hacia el surco mayor. La formación de aductos desencadena una secuencia de eventos que pueden afectar la actividad subsiguiente de las proteínas de unión al ADN, incluidos algunos factores. No se identificaron diferencias clínicamente significativas en la farmacocinética de la lurbinectedina en función de la edad (18 a 85 ańos), el sexo, el peso corporal (39 a 154 kg), la insuficiencia renal de leve a moderada (CLcr 30 a 89 ml/min) o en la insuficiencia hepática leve (bilirrubina total ≤ ULN y AST >ULN, o en la bilirrubina total entre 1 y 1.5 × ULN y en cualquier AST). No se han estudiado los efectos de la insuficiencia renal grave (CLcr <30 ml/min) ni la insuficiencia hepática moderada o grave (bilirrubina total >1.5 × ULN y cualquier AST) en la farmacocinética de la lurbinectedina. Estudios de interacciones farmacológicas: Estudios clínicos: Efectos de los inhibidores de CYP3A en la lurbinectedina: La administración conjunta de itraconazol (un inhibidor potente de CYP3A) aumentó la exposición generalizada (AUC) a la lurbinectedina total en una proporción de 2.7 y a la lurbinectedina sin unir en una proporción de 2.4. Efectos de los inductores de CYP3A en la lurbinectedina: La administración conjunta de bosentán (un inductor moderado de CYP3A) disminuyó la exposición generalizada (AUC) a la lurbinectedina en un 20 % y a la lurbinectedina sin unir en un 19 %. No se considera que estos cambios sean clínicamente relevantes. Estudios in vitro: Enzimas del citocromo P450 (CYP): La lurbinectedina se metaboliza mediante el CYP3A4. La lurbinectedina no es un inhibidor de CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 ni de CYP3A4. La lurbinectedina no es un inductor de CYP1A2 ni de CYP3A4. Sistema de transportadores: La lurbinectedina es un sustrato de MDR1, pero no de OATB1P1, OATP1B3, OCT1 ni de MATE1. La lurbinectedina inhibe al MDR1, OATP1B1, OATP1B3 y al OCT1, pero no al BCRP, BSEP, MATE1, OAT1, OAT3 ni al OCT2.
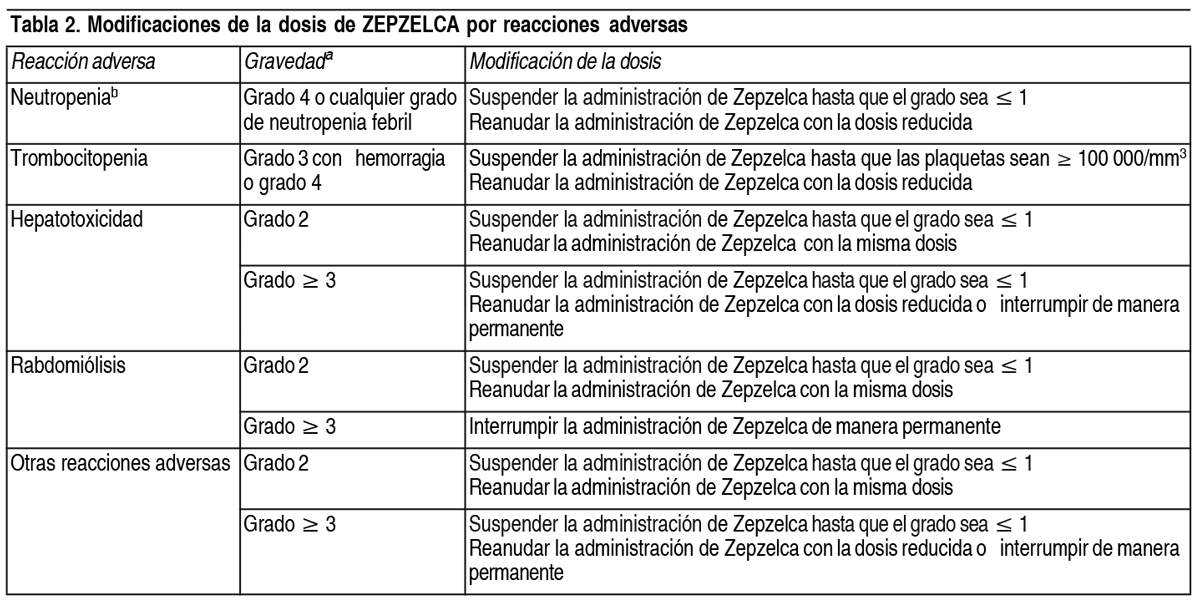
Posología:Dosis recomendada: La dosis recomendada de Zepzelca es de 3.2 mg/m2 por infusión intravenosa durante 60 minutos cada 21 días hasta la evolución de la enfermedad o la toxicidad inaceptable. Se deberá iniciar el tratamiento con Zepzelca solo si el recuento absoluto de neutrófilos (ANC) es de 1500 células/mm3 y el recuento de plaquetas es ≥ 100 000/mm3, como mínimo. Modificación de la dosis por reacciones adversas: Las reducciones de la dosis recomendada por reacciones adversas se detallan en la Tabla 1. Se deberá discontinuar Zepzelca de manera permanente en pacientes que no puedan tolerar 2 mg/m2 o requieran un retraso superior a 2 semanas en la administración de la dosis.
Premedicación: Se deberá considerar administrar los siguientes medicamentos antes de la infusión para la profilaxis antiemética: Corticosteroides (dexametasona de 8 mg por vía intravenosa o equivalente). Antagonistas de la serotonina (ondansetrón de 8 mg por vía intravenosa o equivalente). Preparación, administración y almacenamiento: Zepzelca es un fármaco citotóxico. Se deberán seguir los procedimientos especiales y aplicables de manejo y eliminación. Preparación: Para inyección: 4 mg de lurbinectedina como un polvo liofilizado estéril, sin conservantes, de color blanco a blanquecino, en un vial de dosis única para la reconstitución antes de la infusión intravenosa. Inyectar 8 ml de agua estéril para inyección en el vial, se forma una solución que contiene 0.5 mg/ml de lurbinectedina. Agitar el vial hasta que la disolución se haya completado. Inspeccionar visualmente la solución para comprobar si se observa material particulado y decoloración. La solución reconstituida es transparente, incolora o ligeramente amarillenta, esencialmente sin partículas visibles. Calcular el volumen requerido de la solución reconstituida de la siguiente manera:
Para realizar la administración mediante una línea venosa central, se deberá retirar la cantidad adecuada de solución reconstituida del vial y agregarla en un recipiente de infusión con al menos 100 ml de diluyente (inyección de cloruro de sodio USP al 0.9 % o inyección de glucosa USP al 5 %). Para realizar la administración mediante una línea venosa periférica, se deberá retirar la cantidad adecuada de solución reconstituida del vial y agregarla en un recipiente de infusión con al menos 250 ml de diluyente. Modificación de la dosis para el uso con inhibidores potentes de CYP3A: Se deberá evitar la administración conjunta de Zepzelca con inhibidores potentes de CYP3A. Si no se puede evitar la administración conjunta, se deberá disminuir la dosis de Zepzelca al 50 %. Una vez interrumpida la administración del inhibidor potente de CYP3A por 5 semividas del inhibidor, se deberá aumentar la dosis de Zepzelca hasta la dosis administrada antes de comenzar con la administración del inhibidor.
Modo de Empleo:Manipulaión y eliminación: Zepzelca es un fármaco citotóxico y, al igual que con otros compuestos potencialmente tóxicos, debe tenerse precaución al manipular y preparar soluciones de lurbinectedina. Deben aplicarse los procedimientos especiales para la manipulación y eliminación correctas de medicamentos contra el cáncer. Si la lurbinectedina reconstituida entra en contacto con la piel, la zona deberá lavarse inmediatamente y a fondo con agua y jabón. Si entra en contacto con membranas mucosas, debe lavarse a fondo con agua. Los desechos deben colocarse en recipientes de paredes rígidas para su posterior incineración. Nunca deberán arrojarse medicamentos ni desechos citotóxicos a la red local o desagües.
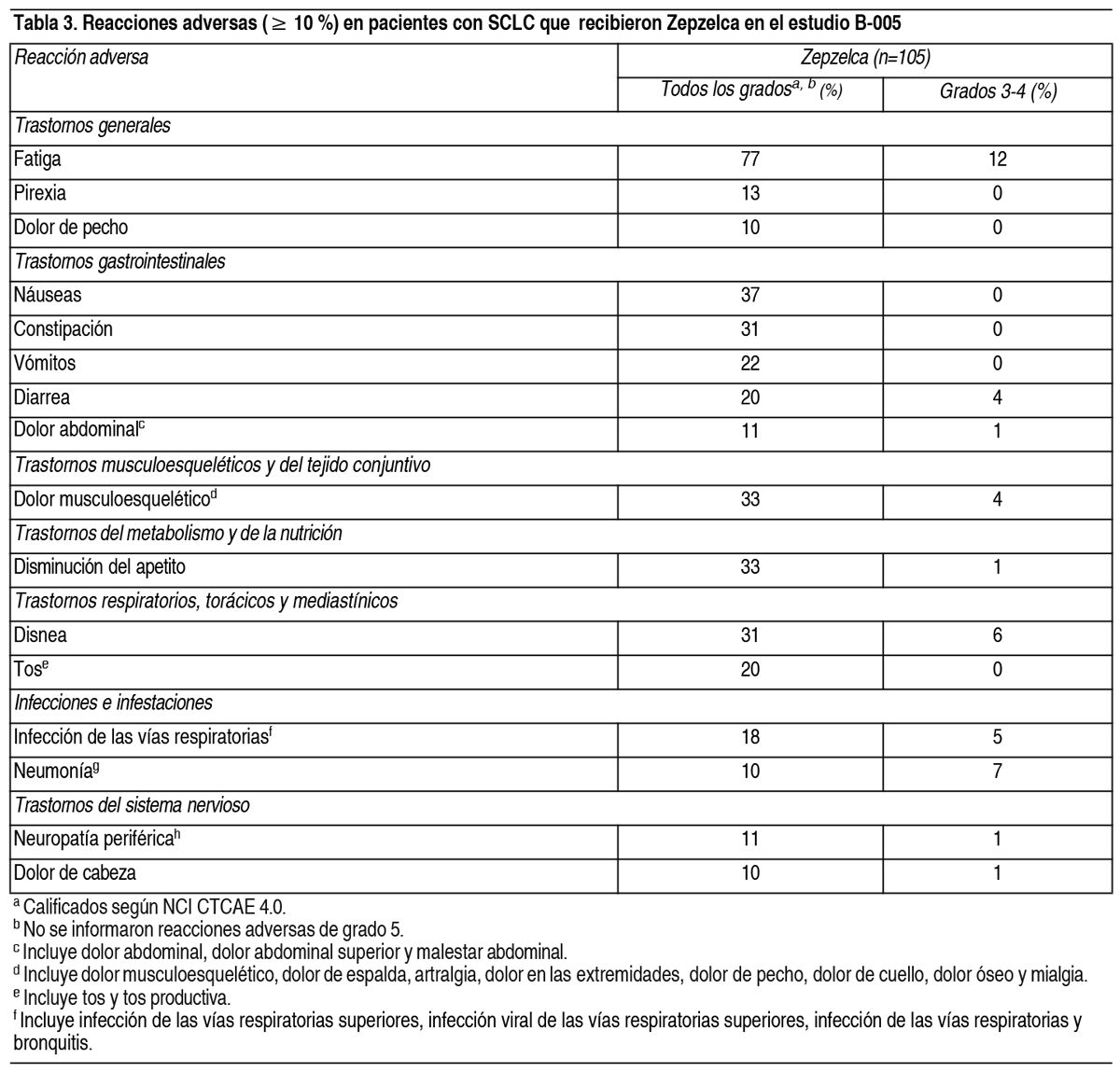
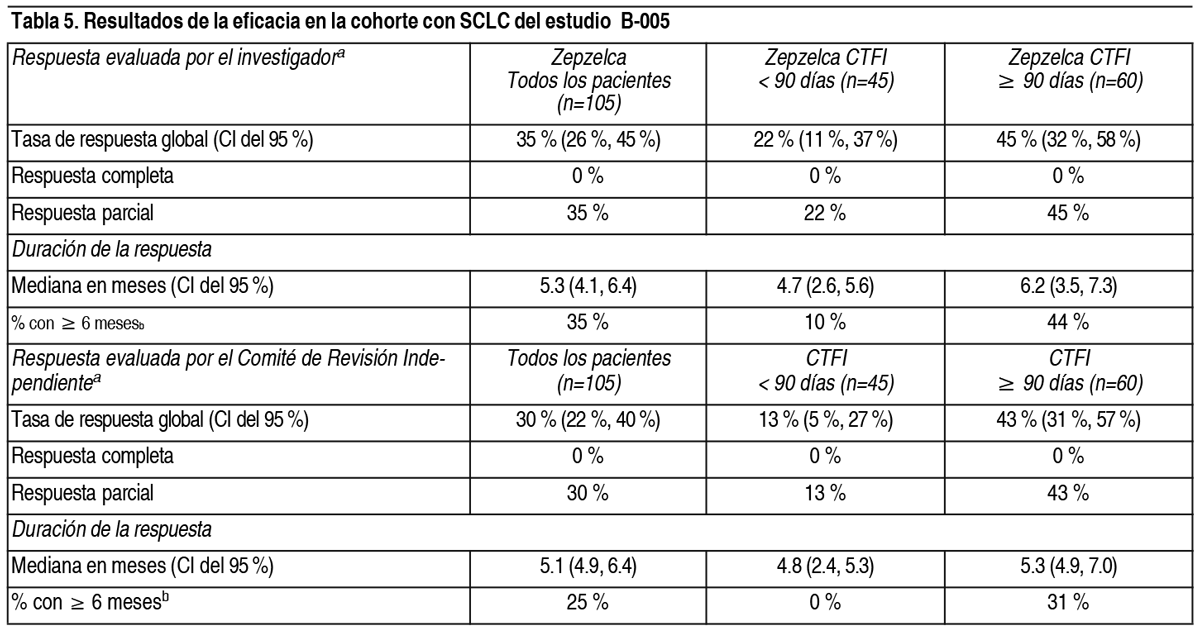
Efectos Colaterales:Las siguientes reacciones adversas clínicamente significativas se describen en otra parte del prospecto: Mielosupresión. Hepatotoxicidad. Extravasación con necrosis tisular. Rabdomiólisis. Experiencia en ensayos clínicos: Dado que los ensayos clínicos se realizan en condiciones muy diferentes, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en ensayos clínicos de otro fármaco y puede que no reflejen las tasas observadas en la práctica. El pool de la población de seguridad descrito en la sección Advertencias y precauciones refleja la exposición al Zepzelca como un agente único administrado con una dosis de 3.2 mg/m2 por vía intravenosa cada 21 días en 554 pacientes con tumores sólidos avanzados. Entre los 554 pacientes que recibieron Zepzelca, incluidos los 105 pacientes con carcinoma de células pequeńas de pulmón (SCLC) en PM1183-B-005-14 (estudio B-005), el 24 % estuvo expuesto durante 6 meses o más y el 5 % estuvo expuesto durante más de 1 ańo. Cáncer de pulmón de células pequeńas (SCLC): Se evaluó la seguridad de Zepzelca en una cohorte de 105 pacientes con SCLC tratados previamente en el estudio B-005. Los pacientes recibieron una dosis de 3.2 mg/m2 de Zepzelca por vía intravenosa cada 21 días. Todos los pacientes de este estudio recibieron una pauta posológica antiemética prestablecida que consistía en un corticosteroide y un antagonista de la serotonina. Los pacientes podían recibir el G-CSF en la profilaxis secundaria (es decir, después de que los pacientes hayan tenido una disminución inicial en el recuento de glóbulos blancos), pero no en la profilaxis primaria. Entre los pacientes que recibieron Zepzelca, el 29 % estuvo expuesto durante 6 meses o más y el 6 % estuvo expuesto durante más de 1 ańo. Las reacciones adversas graves se produjeron en el 34 % de los pacientes que recibieron Zepzelca. Las reacciones adversas graves en ≥ 3 % de los pacientes incluyeron neumonía, neutropenia febril, neutropenia, infección de las vías respiratorias, anemia, disnea y trombocitopenia. La interrupción permanente debido a una reacción adversa se produjo en dos pacientes (1.9 %) que recibieron Zepzelca. Las reacciones adversas provocaron la interrupción permanente en ≥ 1 % que recibió Zepzelca, incluidas la neuropatía periférica y la mielosupresión. Las interrupciones de la dosis debido a una reacción adversa se produje- ron en el 30.5 % de los pacientes que recibieron Zepzelca. Las reacciones adversas que requirieron la interrupción de la dosis en ≥ 3 % de los pacientes que recibieron Zepzelca incluyeron neutropenia e hipoalbuminemia. Las reducciones de la dosis debido a una reacción adversa se produjeron en el 25 % de los pacientes que recibieron Zepzelca. Las reacciones adversas que requirieron reducciones de la dosis en ≥ 3 % de los pacientes que recibieron Zepzelca incluyeron neutropenia, neutropenia febril y fatiga. Las reacciones adversas más frecuentes, incluidos los valores anormales de laboratorio, (≤20 %) fueron: Leucopenia, fatiga, anemia, neutropenia, aumento de la creatinina, aumento de la alanina aminotransferasa, aumento de la glucosa, trombocitopenia, náuseas, disminución del apetito, dolor musculo-esquelético, disminución de la albúmina, constipación, disnea, disminución del sodio, aumento de la aspartato aminotransferasa, vómitos, tos, disminución del magnesio y diarrea. En la Tabla 3, se resumen las reacciones adversas en la cohorte con SCLC del estudio B-005.
Las reacciones adversas clínicamente relevantes en < 10 % de los pacientes que recibieron Zepzelca incluyen disgeusia, neutropenia febril y neumonitis. En la Tabla 4, se resumen los valores anormales de laboratorio del estudio B-005.
Experiencia posterior a la comercialización: Se identificaron las siguientes reacciones adversas durante el uso posterior a la aprobación de Zepzelca. Debido a que estas reacciones se informan voluntariamente a partir de una población de tamańo incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco. Trastornos generales y alteraciones en el lugar de administración: Extravasación con necrosis tisular que requiere desbridamiento. Trastornos osteomusculares y del tejido conectivo: Rabdomiólisis. Trastornos metabólicos y alimentarios: Síndrome de lisis tumoral. Estudios clínicos: PM1183-B-005-14 (estudio B-005; NCT02454972) es un estudio multicéntrico, abierto, de cohortes múltiples, en el que se evalúa Zepzelca como un agente único en pacientes con tumores sólidos avanzados o metastásicos. Una cohorte de pacientes con cáncer de pulmón de células pequeńas (SCLC) y progresión de la enfermedad durante o después de la quimioterapia con un derivado del platino recibió una dosis de Zepzelca de 3.2 mg/m2 por infusión intravenosa cada 21 días (1 ciclo). Los pacientes recibieron una mediana de 4 ciclos de Zepzelca (intervalo de 1 a 24 ciclos). En el estudio se excluyeron los pacientes con afectación del sistema nervioso central (SNC), disnea de grado ≥ 3, demanda de oxígeno diaria e intermitente, hepatitis o cirrosis y los pacientes inmunocomprometidos. Se realizaron evaluaciones tumorales cada 6 semanas durante las primeras 18 semanas y cada 9 semanas en adelante. La medición del resultado de la eficacia principal confirmó la tasa de respuesta global (TRG) evaluada por el investigador. Las mediciones adicionales de los resultados de la eficacia incluyeron la duración de la respuesta (DR) y un Comité de Revisión Independiente (IRC) evaluó la TRG mediante el uso de los criterios de evaluación de la respuesta en tumores sólidos (RECIST v1.1). En total, se enrolaron a 105 pacientes con SCLC que evolucionaron durante o después de la quimioterapia con un derivado del platino. La mediana de edad fue de 60 ańos (intervalo: de 40 a 83) con el 65 % de los pacientes < 65 ańos y el 35 % de los pacientes ≤ 65 ańos, y el 60 % eran hombres. La mayoría de los pacientes (75 %) eran blancos, el 1 % era asiático, el 1 % era negro y el 23 % no se informaron. El estado de performance del ECOG al inicio fue de 0 o 1 en el 92 % de los pacientes, y el 92 % eran exfumadores/fumadores actuales. Todos los pacientes recibieron al menos 1 línea de quimioterapia con un derivado del platino (intervalo de 1 a 2 líneas) y la radioterapia se había administrado previamente en el 71 % de los pacientes. 8 pacientes (8 %) recibieron inmunoterapia previamente además de la quimioterapia con un derivado del platino. 60 pacientes (57 %) tuvieron SCLC sensible al platino, definida como la recaída o progresión ≥ 90 días después de la última dosis de tratamiento con platino (intervalo sin quimioterapia [CTFI] ≥ 90 días). Los 45 pacientes restantes tuvieron SCLC resistente al platino, definida como la recaída o progresión < 90 días después de la última dosis de tratamiento con platino (CTFI < 90 días). En la Tabla 5, se resumen las medidas de eficacia clave evaluadas por el investigador y por el Comité de Revisión Independiente en todos los pacientes y en los subgrupos resistentes y sensibles al platino
Información de consulta del paciente: Recomendar a los pacientes que lean la Información para el paciente. Mielosupresión: Se deberá recomendar a los pacientes que se comuniquen con su médico de inmediato en caso de presentar fiebre, otros signos de infección, moretones poco frecuentes, hemorragia, cansancio o palidez. Hepatotoxicidad: Se deberá recomendar a los pacientes que se comuniquen con su médico de inmediato en caso de presentar signos y síntomas que sugieran hepatotoxicidad. Extravasación con necrosis tisular: Se deberá recomendar a los pacientes que consulten con el médico de inmediato en caso de signos y síntomas de extravasación. El inicio de la necrosis después de la extravasación puede variar. Rabdomiólisis: Se deberá recomendar a los pacientes que consulten con el médico de inmediato en caso de signos y síntomas de rabdomiólisis. Toxicidad embriofetal: Se deberá informar a las mujeres embarazadas y a las mujeres en edad reproductiva sobre el posible riesgo al feto. Se deberá recomendar a las mujeres que le informen a su médico sobre un embarazo conocido o sospecha de embarazo. Se deberá recomendar a las mujeres en edad reproductiva que utilicen un método anticonceptivo eficaz durante el tratamiento con Zepzelca y durante 6 meses después de la última dosis. Se deberá recomendar a los hombres con parejas mujeres en edad reproductiva que utilicen un método anticonceptivo eficaz durante el tratamiento con Zepzelca y durante 4 meses después de la última dosis. Lactancia: Se deberá recomendar a las mujeres que no amamanten durante el tratamiento con Zepzelca y durante al menos 2 semanas después de la última dosis. Interacciones farmacológicas: Se deberá recomendar a los pacientes que le informen a su médico sobre todos los medicamentos concomitantes y los suplementos herbales y alimenticios. Se deberá recomendar a los pacientes que eviten el consumo de productos con pomelo y naranja agria durante el tratamiento con Zepzelca.
Contraindicaciones: Hipersensibilidad al principio activo lurbinectedina o a alguno de los excipientes (consultar la fórmula).
Advertencias:Mielosupresión: Zepzelca puede causar mielosupresión. En estudios clínicos realizados en 554 pacientes con tumores sólidos avanzados que recibieron Zepzelca, se produjo neutropenia de grado 3 o 4 en el 41 % de los pacientes, con una mediana de tiempo transcurrido hasta la aparición de 15 días y una mediana de duración de 7 días. Se produjo neutropenia febril en el 7 % de los pacientes. Se produjo sepsis en el 2 % de los pacientes y fue mortal en el 1 % (todos los casos se produjeron en pacientes con tumores sólidos además del SCLC). Se produjo trombocitopenia de grado 3 o 4 en el 10 % de los pacientes, con una mediana de tiempo transcurrido hasta la aparición de 10 días y una mediana de duración de 7 días. Se produjo anemia de grado 3 o 4 en el 17 % de los pacientes. Se deberá administrar Zepzelca solo a los pacientes con un recuento de neutrófilos de 1500 células/mm3 y un recuento de plaquetas ≥ 100 000/mm3, como mínimo. Se deberán monitorear los hemogramas, incluidos el recuento de neutrófilos y plaquetas antes de cada administración. En caso de que el recuento de neutrófilos sea menor a 500 células/mm3 o de cualquier valor menor al límite inferior de lo normal, se recomienda utilizar el G-CSF. Se deberá suspender, reducir la dosis o discontinuar de forma permanente la administración de Zepzelca en función de la gravedad. Extravasación con necrosis tisular: Puede ocurrir la extravasación de Zepzelca, que puede causar lesiones de la piel y del tejido blando, incluso necrosis que requiere desbridamiento. Se deberá considerar el uso de una vía venosa central para disminuir el riesgo de extravasación, particularmente en los pacientes con acceso limitado a las venas. Se deberá controlar que los pacientes no presenten signos y síntomas de extravasación durante la infusión de Zepzelca. En caso de extravasación, se deberá suspender de inmediato la infusión, eliminar la vía de infusión y observar los signos y síntomas de necrosis tisular. El inicio de la necrosis después de la extravasación puede variar. Se deberá administrar un tratamiento paliativo y consultar con el especialista (inyección de cloruro de sodio USP al 0.9 % o inyección de glucosa USP al 5 %). Administración: Se deberán inspeccionar los productos terminados parenterales para comprobar si se observa material particulado o decoloración antes de la administración, siempre y cuando lo permitan la solución y el recipiente. Si se observa material particulado, no se deberá administrar el fármaco. Se puede administrar Zepzelca con o sin un filtro incorporado. Si se utilizan vías de infusión con filtros incorporados para administrar Zepzelca, se recomiendan los de polietersulfona (PES) con tamańos de poro de 0.22 micrómetros. No se deben utilizar filtros incorporados de membrana de nailon cuando la solución reconstituida de Zepzelca se diluye con cloruro de sodio inyectable al 0.9 % de la USP. Se observó la adsorción de Zepzelca a los filtros de membrana de nailon cuando se utiliza cloruro de sodio inyectable al 0.9 % de la USP como diluyente. Se demostró la compatibilidad con otros materiales de administración intravenosa y la solución diluida de Zepzelca en los siguientes materiales: Envases de poliolefina (polietileno, polipropileno y mezclas). Equipos de infusión de policloruro de vinilo (PVC) (que no contienen DEHP), poliuretano y poliolefina (polietileno, polipropileno y polibutadieno). Sistemas de acceso venoso implantables con puertos de titanio y resina plástica, y con catéteres intravenosos de poliuretano o silicona. No se deberán administrar Zepzelca y otros medicamentos intravenosos en simultáneo con la misma vía intravenosa. Almacenamiento de la solución de infusión: Si no se utiliza inmediatamente después de la reconstitución o dilución, la solución de Zepzelca puede almacenarse antes de su administración durante un máximo de 24 horas después de la reconstitución, incluido el tiempo de infusión, a temperatura ambiente/con luz ambiental o en condiciones de refrigeración de 2 °C a 8 °C.
Precauciones:Efecto de otros fármacos en Zepzelca: Inhibidores potentes y moderados de CYP3A: La administración conjunta de Zepzelca con un inhibidor potente o moderado de CYP3A aumenta la exposición sistémica al lurbinectedina, lo que puede aumentar la incidencia y gravedad de las reacciones adversas al Zepzelca. Se deberá evitar el consumo de pomelos y naranjas agrias durante el tratamiento con Zepzelca, dado que contienen inhibidores potentes o moderados de CYP3A. Inhibidores potentes de CYP3A: Se deberá evitar la administración conjunta de Zepzelca con inhibidores potentes de CYP3A. Si no es posible evitarlo, se deberá disminuir la dosis de Zepzelca. Inhibidores moderados de CYP3A: Se deberá evitar la administración conjunta de Zepzelca con inhibidores moderados de CYP3A. Si no es posible evitarlo, se deberá disminuir la dosis de Zepzelca, si se indica clínicamente. Inductores potentes de CYP3A: Se deberá evitar la administración conjunta de Zepzelca con un inductor potente de CYP3A. Esto podría disminuir la exposición sistémica a la lurbinectedina, lo que puede reducir la eficacia de Zepzelca. Uso en poblaciones específicas: Embarazo: Resumen del riesgo: En base a datos obtenidos en animales y a su mecanismo de acción, Zepzelca puede causar dańo al feto cuando se administra en una mujer embarazada. No hay datos disponibles para informar sobre el riesgo del para determinar los signos y síntomas de extravasación. Las infusiones posteriores se deberán administrar en un lugar de administración que no haya estado afectado por la extravasación. Rabdomiólisis: Se observó rabdomiólisis en pacientes tratados con Zepzelca. Se deberán controlar los niveles de creatina fosfocinasa (CPK) antes de comenzar con la administración de Zepzelca y periódicamente durante el tratamiento, por indicación del médico. Se deberá suspender o reducir la dosis en función de la gravedad. Hepatotoxicidad: Zepzelca puede causar hepatotoxicidad. En estudios clínicos realizados en 554 pacientes con tumores sólidos avanzados que recibieron Zepzelca, se observaron aumentos de grado 3 de la ALT y la AST en el 6 % y 3 % de los pacientes, respectivamente; y aumentos de grado 4 de la ALT y la AST en el 0.4 % y 0.5 % de los pacientes, respectivamente. La mediana de tiempo transcurrido hasta la aparición del aumento de grado ≥ 3 en las transaminasas fue de 8 días (intervalo: de 3 a 49), con una mediana de la duración de 7 días. Se deberán monitorear los análisis de función hepática antes de iniciar la administración de Zepzelca, de forma periódica durante el tratamiento y como se indica clínicamente. Se deberá suspender, reducir la dosis o discontinuar de forma permanente la administración de Zepzelca en función de la gravedad. Toxicidad embriofetal: En base a datos obtenidos en animales y a su mecanismo de acción, Zepzelca puede dańar al feto cuando se administra en una mujer embarazada. La administración intravenosa de una dosis única de lurbinectedina (aproximadamente 0.2 veces la dosis clínica de 3.2 mg/m2) en animales preńados durante el período de organogénesis causó el 100 % de la embrioletalidad en ratas. Se deberá informar a las mujeres embarazadas sobre el posible riesgo al feto. Mujeres: Se deberá recomendar a las pacientes mujeres en edad reproductiva que utilicen un método anticonceptivo eficaz durante el tratamiento con Zepzelca y durante 6 meses después de la última dosis. Hombres: Se deberá recomendar a los hombres con una pareja sexual mujer en edad reproductiva que utilicen un método anticonceptivo eficaz durante el tratamiento con Zepzelca y durante 4 meses después de la última dosis. Uso pediátrico: No se ha establecido la seguridad y la eficacia de Zepzelca en pacientes pediátricos. Uso geriátrico: De los 105 pacientes con SCLC a los que se les administró Zepzelca en estudios clínicos, 37 pacientes (35 %) eran mayores de 65 ańos, mientras que 9 pacientes (9 %) eran mayores de 75 ańos. No se observó ninguna diferencia general en la eficacia entre los pacientes mayores de 65 ańos y los pacientes más jóvenes. Hubo una mayor incidencia de reacciones adversas graves en pacientes ≥ 65 ańos que en pacientes < 65 ańos (el 49 % en comparación con el 26 %, respectivamente). Las reacciones adversas graves informadas con mayor frecuencia en pacientes ≥ 65 ańos se relacionaron con la mielosupresión y fueron: Neutropenia febril (11 %), neutropenia (11 %), trombocitopenia (8 %) y anemia (8 %). Insuficiencia hepática: No se ha estudiado el efecto de la insuficiencia hepática moderada o grave (bilirrubina total > 1.5 × ULN y cualquier AST) en la farmacocinética del lurbinectedina. No se recomienda realizar un ajuste de la dosis de Zepzelca en pacientes con insuficiencia hepática leve (bilirrubina total ≤ ULN y AST > ULN o bilirrubina total de 1 a 1,5 × ULN y cualquier AST). Carcinogénesis, mutagénesis, trastornos de la fertilidad: No se ha realizado el análisis de la carcinogenicidad de lurbinectedina. La lurbinectedina es genotóxica para las células de mamíferos en presencia uso de Zepzelca en mujeres embarazadas. La administración intravenosa de una dosis única de lurbinectedina (aproximadamente 0.2 veces la dosis clínica de 3.2 mg/m2) en ratas preńadas durante el período de organogénesis causó embrioletalidad. Se deberá informar a las mujeres embarazadas sobre el posible riesgo al feto. Se desconoce el riesgo de fondo previsto de los defectos congénitos principales y del aborto espontáneo en la población indicada. Todos los embarazos presentan un riesgo de fondo de defecto congénito, pérdida u otros desenlaces adversos. En la población general de los EE. UU., el riesgo de fondo previsto de los efectos congénitos principales y del aborto espontáneo en embarazos clínicamente reconocidos es del 2 al 4 % y del 15 al 20 %, respectivamente. Datos: Datos en animales: En un estudio de toxicidad reproductiva, la administración de una dosis única de 0.6 mg/m2 de lurbinectedina (aproximadamente 0.2 veces de la dosis en humanos de 3.2 mg/m2) en ratas preńadas el día de gestación 10 provocó el 100 % de la pérdida posimplantación. Lactancia: Resumen de los riesgos: No hay datos sobre la presencia de lurbinectedina en la leche humana o sus efectos en el lactante ni en la producción de leche. Dada la posibilidad de que se produzcan reacciones adversas graves en lactantes, se deberá recomendar a las mujeres que no amamanten durante el tratamiento con Zepzelca ni durante 2 semanas después de la última dosis. Mujeres y hombres con capacidad reproductiva: Zepzelca puede causar embrioletalidad con dosis más bajas que la dosis de 3.2 mg/m2 administrada en humanos. Prueba de embarazo: Se deberá verificar el estado de embarazo de las mujeres en edad reproductiva antes de iniciar el tratamiento con Zepzelca. Anticoncepción y ausencia de la activación metabólica. La lurbinectedina no fue mutagénico in vitro en el ensayo de mutación inversa bacteriana (Ames). No se realizaron estudios de fertilidad con lurbinectedina. No hubo hallazgos en los órganos reproductivos en los estudios generales de toxicología en ratas, perros o monos; sin embargo, todas las dosis máximas y las exposiciones en estos estudios estuvieron en los niveles más bajos que las de la dosis de 3.2 mg/m2 administrada en humanos.
Sobredosificación:Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los centros de toxicología: Hospital de Pediatría «Ricardo Gutiérrez» Teléfono: (011) 4962 6666 / 2247. Hospital «A. Posadas» Teléfono: (011) 4654 6648 / 4658 7777. Centro de Asistencia Toxicológica de La Plata, Teléfono: (0221) 451 5555.
Conservación: Almacenar refrigerado de 2 °C a 8 °C.
Observaciones: Mantener fuera del alcance de los nińos. Este medicamento solo debe utilizarse bajo estricto control y vigilancia médica y no puede repetirse sin nueva receta.
Presentaciones: Envase conteniendo 1 vial de dosis única.
 Laboratorio
Laboratorio