Oncológicos y Terapias Relacionadas: Otros Agentes Antineoplásicos
Información farmacológica
Composición: Cada ml contiene: Ramucirumab 10 mg, Glicina 9.98 mg, L-Histidina 0.65 mg, Monoclorhidrato de L-Histidina 1.22 mg, Polisorbato 80 0.10 mg, Cloruro de Sodio 4.38 mg, Agua para Inyección c.s.p. 1 ml. Lista de excipientes: Glicina, L-Histidina, Monoclorhidrato de L-Histidina, Polisorbato 80, Cloruro de Sodio, Agua para Inyección c.s.p.
Descripción: Cyramza® es una solución inyectable estéril, sin conservantes, de transparente a ligeramente opalescente e incolora a ligeramente amarilla, con pH entre 5.7 y 6.3 y sin partículas visibles para perfusión intravenosa una vez diluida y preparada. Cyramza® se presenta en viales unidosis de 100 mg de ramucirumab en 10 ml (10 mg/ml) o 500 mg de ramucirumab en 50 ml (10 mg/ml).
Acción Terapéutica:Grupo farmacoterapéutico: Agentes antineoplásicos, inhibidores del VEGF/VEGFR (factor de crecimiento del endotelio vascular). Código ATC: L01FG02.
Indicaciones:Cáncer gástrico: Cyramza® en combinación con paclitaxel está indicado para el tratamiento de pacientes adultos con cáncer gástrico avanzado o adenocarcinoma de la unión gastroesofágica con progresión de la enfermedad tras quimioterapia previa con platino y fluoropirimidina (ver sección Propiedades Farmacodinámicas). Cyramza® en monoterapia está indicado para el tratamiento de pacientes adultos con cáncer gástrico avanzado o adenocarcinoma de la unión gastroesofágica con progresión de la enfermedad tras quimioterapia previa con platino o fluoropirimidina, para quienes el tratamiento en combinación con paclitaxel no es apropiado (ver sección Propiedades Farmacodinámicas). Cáncer colorrectal: Cyramza® en combinación con FOLFIRI (irinotecán, ácido folínico y 5-fluorouracilo), está indicado para el tratamiento de pacientes adultos con cáncer colorrectal metastásico (metastatic colorectal cancer - mCRC, por sus siglas en inglés) con progresión de la enfermedad durante o tras terapia previa con bevacizumab, oxaliplatino y una fluoropirimidina. Cáncer de pulmón no microcítico: Cyramza® en combinación con erlotinib está indicado como tratamiento en primera línea de pacientes adultos con cáncer de pulmón no microcítico metastásico con mutaciones activadoras del receptor del factor de crecimiento epidérmico (EGFR) (ver sección Propiedades Farmacodinámicas). Cyramza® en combinación con docetaxel está indicado para el tratamiento de pacientes adultos con cáncer de pulmón no microcítico localmente avanzado o metastásico con progresión de la enfermedad tras quimioterapia basada en platino. Carcinoma hepatocelular: Cyramza® en monoterapia está indicado para el tratamiento de pacientes adultos con carcinoma hepatocelular avanzado o no resecable que tienen una alfafetoproteína (AFP) ≥ 400 ng/ml y que han sido previamente tratados con sorafenib.
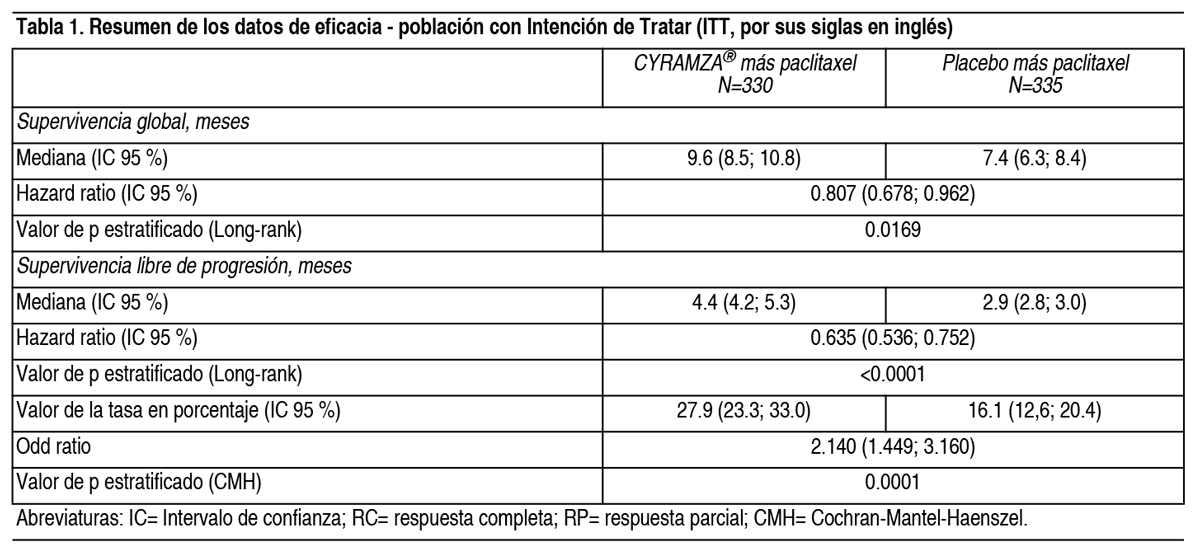
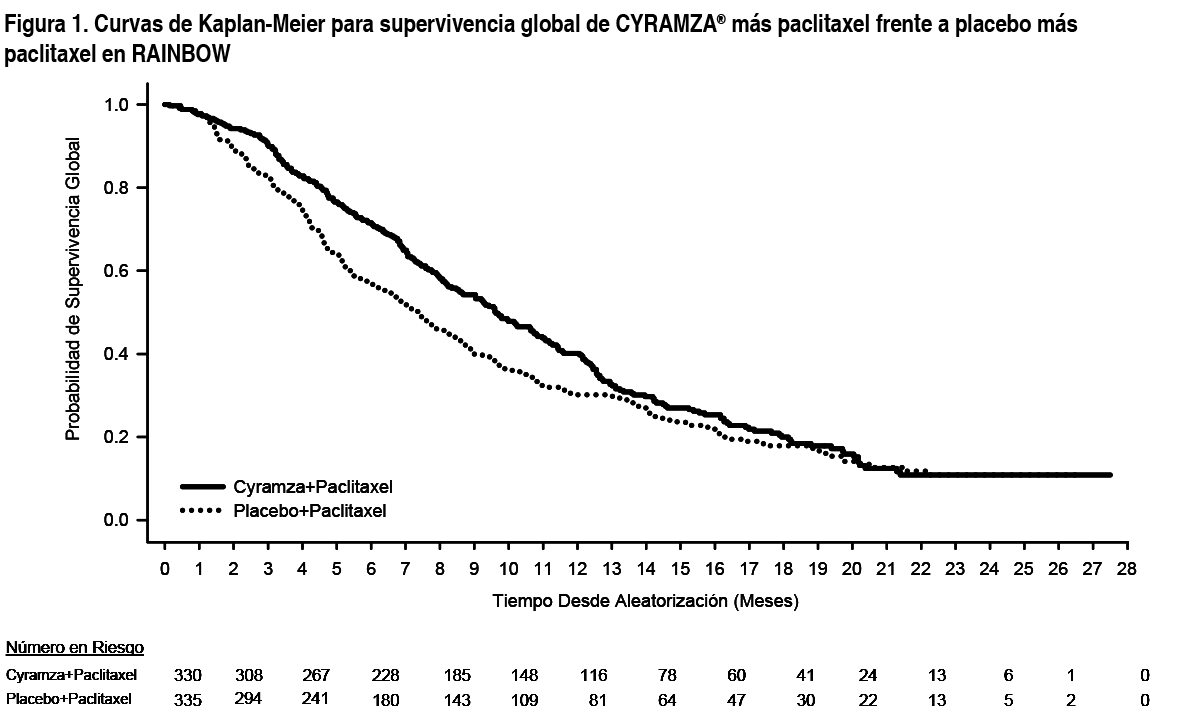
Propiedades:Propiedades farmacodinámicas: Mecanismo de acción: El receptor tipo 2 del factor de crecimiento del endotelio vascular (Vascular Endothelial Growth Factor - VEGF, por sus siglas en inglés) es el mediador clave de la angiogénesis inducida por el VEGF. Ramucirumab es un anticuerpo humano dirigido a receptores que se une específicamente al Receptor 2 del VEGF bloqueando la unión de los ligandos VEGF-A, VEGF-C y VEGF-D. Como resultado, ramucirumab inhibe la activación ligando-dependiente del Receptor 2 de VEGF y sus componentes posteriores de la cascada de seńalización, incluyendo las proteínas kinasas activadas por mitógeno p44/p42, la proliferación ligando inducida y la migración de las células endoteliales humanas. Eficacia clínica y seguridad: Cáncer gástrico: RAINBOW: RAINBOW es un ensayo clínico global, aleatorizado, doble ciego de Cyramza® más paclitaxel frente a placebo más paclitaxel, llevado a cabo en 665 pacientes con cáncer localmente recurrente e irresecable o cáncer gástrico metastásico (incluyendo adenocarcinoma de la unión gastroesofágica GEJ, por sus siglas en inglés) tras quimioterapia con platino y fluoropirimidina, con o sin antraciclinas. La variable primaria fue supervivencia global (overall survival - OS, por sus siglas en inglés) y las variables secundarias incluyeron supervivencia libre de progresión (progression-free survival - PFS, por sus siglas en inglés) y la tasa de respuesta global (overall response rate - ORR, por sus siglas en inglés). Se requería que los pacientes hubiesen presentado progresión de la enfermedad durante el tratamiento en primera línea o dentro de los 4 meses tras la última dosis del mismo y tuvieran ECOG PS (pacientes con estado funcional según la escala Eastern Cooperative Oncology Group) 0-1. Los pacientes fueron asignados de forma aleatoria en un ratio 1:1 para recibir Cyramza® más paclitaxel (n= 330) o placebo más paclitaxel (n= 335). La aleatorización se estratificó por región geográfica, tiempo a la progresión desde el comienzo del tratamiento en primera línea (< 6 meses frente ≥ 6 meses) y cuantificación de la enfermedad. Tanto Cyramza® 8 mg/kg como placebo fueron administrados por perfusión intravenosa cada 2 semanas (los días 1 y 15) de un ciclo de 28 días. Paclitaxel 80 mg/m2 se administró por perfusión intravenosa los días 1, 8 y 15 de cada ciclo de 28 días. La mayoría (75 %) de los pacientes aleatorizados en el estudio, recibieron previamente una terapia combinada de platino y fluoropirimidina sin antraciclina. El resto (25 %), recibieron previamente una terapia combinada de platino y fluoropirimidina con antraciclina. Dos tercios de los pacientes mostraron progresión de la enfermedad, cuando aún se encontraban en tratamiento de primera línea (66.8 %). Las características basales demográficas y de la enfermedad estaban globalmente balanceadas entre los grupos del estudio: La mediana de edad fue 61 ańos; 71 % de pacientes eran hombres; 61 % eran caucásicos, 35 % asiáticos; ECOG PS fue 0 para el 39 % de los pacientes, 1 para el 61 %; 81 % de los pacientes tenía enfermedad cuantificable y el 79 % presentaba cáncer gástrico, 21 % presentaba adenocarcinoma de GEJ. La mayoría de los pacientes (76 %) había presentado progresión de la enfermedad durante los 6 meses desde el comienzo del tratamiento en primera línea. Para los pacientes tratados con Cyramza® más paclitaxel, la mediana de la duración del tratamiento fue de 19 semanas y para pacientes tratados con placebo más paclitaxel, la mediana de duración del tratamiento fue de 12 semanas. La mediana de la intensidad de dosis relativa de Cyramza® fue 98.6 % y de placebo fue 99.6 %. La mediana de la intensidad de dosis relativa de paclitaxel fue 87.7 % en el grupo de Cyramza® más paclitaxel y 93.2 % en el grupo de placebo más paclitaxel. Un porcentaje parecido de pacientes interrumpió el tratamiento debido a reacciones adversas: 12 % de los pacientes tratados con Cyramza® más paclitaxel comparado con el 11 % de pacientes tratados con placebo más paclitaxel. En el 47.9 % de los pacientes que recibieron Cyramza® más paclitaxel se administró un tratamiento anticanceroso sistémico tras la interrupción del tratamiento, frente al 46.0 % de los pacientes que recibieron placebo más paclitaxel. La supervivencia global fue mejorada de forma estadísticamente significativa en pacientes que recibieron Cyramza® más paclitaxel comparado con aquellos que recibieron placebo más paclitaxel (Hazard Ratio - HR 0.807; IC 95 %: 0.678 a 0.962; p=0.0169). Hubo un aumento de la mediana de supervivencia de 2.3 meses a favor del grupo de tratamiento de Cyramza® más paclitaxel: 9.63 meses en el brazo de Cyramza® más paclitaxel y 7.36 meses en el grupo de placebo más paclitaxel. La supervivencia libre de progresión mejoró de forma estadísticamente significativa para los pacientes que recibieron Cyramza® más paclitaxel comparada con aquellos que recibieron placebo más paclitaxel (HR 0.635; IC 95 % 0.536 a 0.752; p < 0.0001). Hubo un aumento en la mediana de PFS de 1.5 meses a favor del grupo de tratamiento de Cyramza®: 4.4 meses en el grupo de Cyramza® más paclitaxel y 2.9 meses en el grupo de placebo más paclitaxel. La tasa de respuesta objetiva {ORR [respuesta completa (RC) + respuesta parcial (RP)]} mejoró de forma significativa en pacientes que recibieron Cyramza® más paclitaxel comparado con aquellos que recibieron placebo más paclitaxel (Odds ratio 2.140; IC 95 %: 1.499 a 3.160; p=0.0001). El ORR en el grupo de Cyramza® más paclitaxel fue de 27.9 % y 16.1 % en el grupo de placebo más paclitaxel. En subgrupos predefinidos según edad, sexo, raza se observaron mejoras de forma consistente en OS y PFS, así como en la mayoría de otros subgrupos predefinidos. Los datos de eficacia se muestran en la Tabla 1.
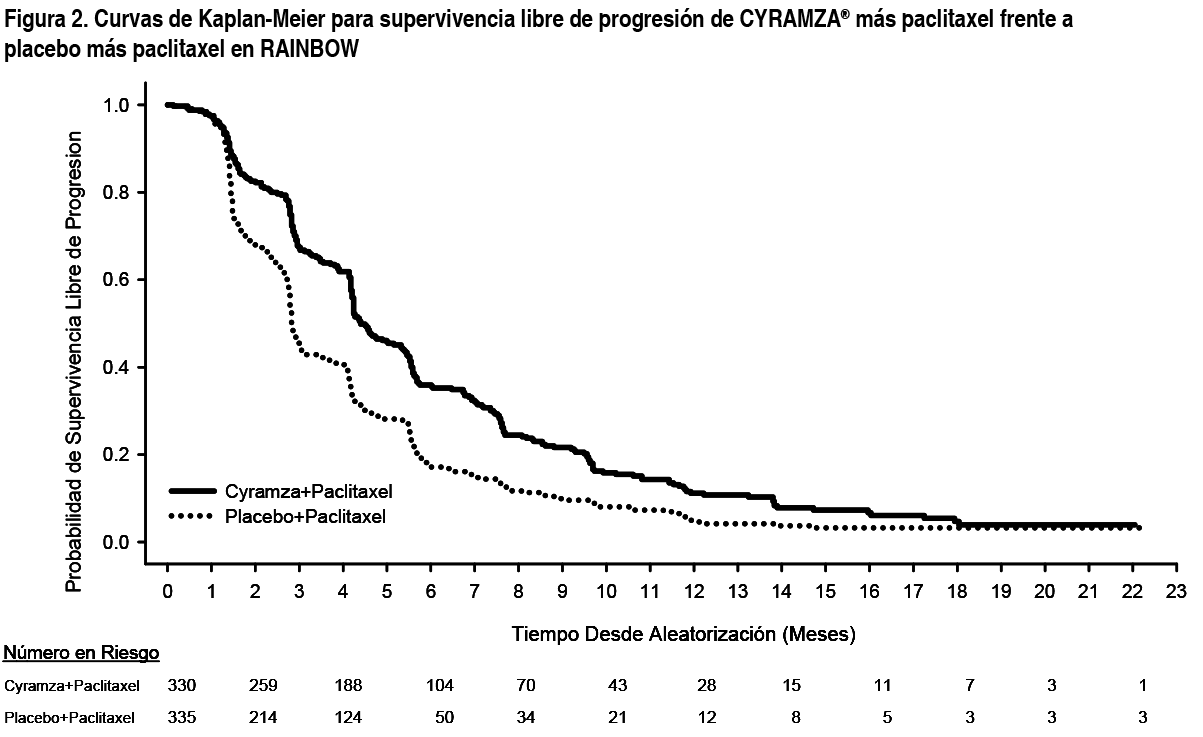
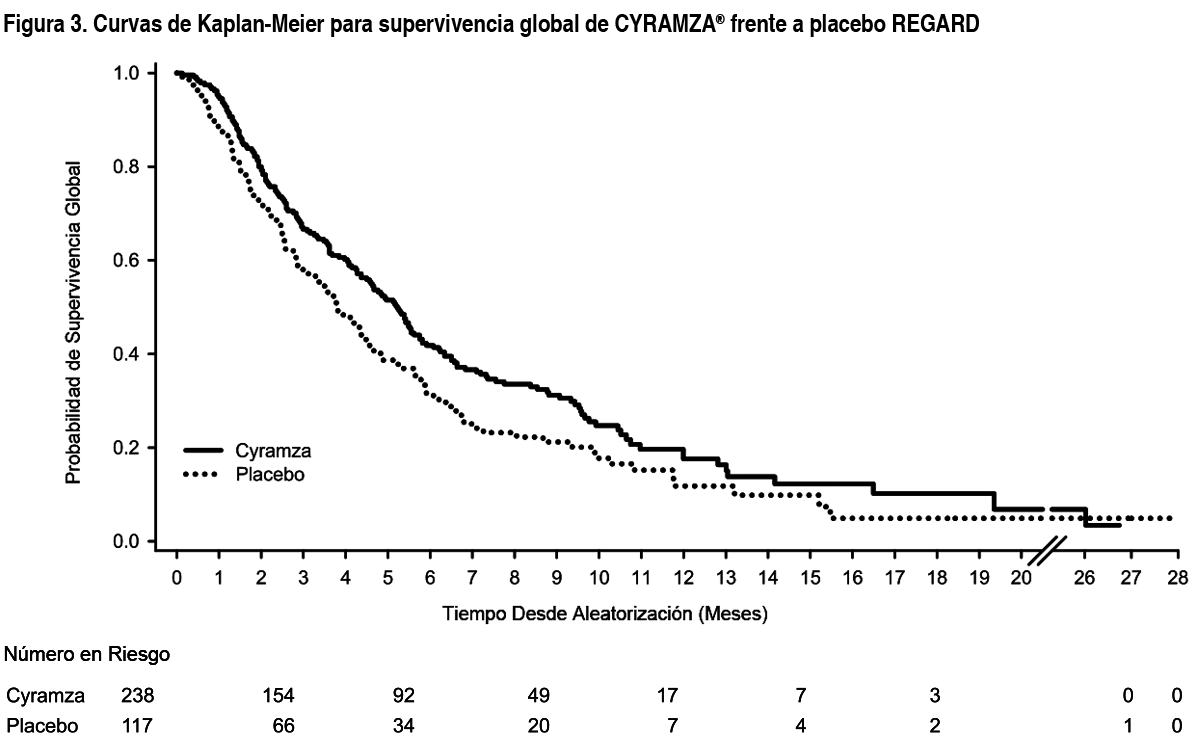
REGARD: REGARD es un ensayo clínico multinacional, aleatorizado, doble ciego de Cyramza® más el mejor tratamiento de soporte (BSC, por sus siglas en inglés) frente a placebo más BSC, llevado a cabo en 355 pacientes con cáncer localmente recurrente e irresecable o cáncer gástrico metastásico (incluyendo adenocarcinoma de GEJ) tras quimioterapia con platino o fluoropirimidina. La variable primaria fue OS y las variables secundarias incluyeron PFS. Se requería que los pacientes hubiesen presentado progresión de la enfermedad durante o dentro de los 4 meses tras la última dosis del tratamiento, en primera línea para la enfermedad metastásica, o durante o dentro de los 6 meses tras la última dosis de la terapia adyuvante y tuvieran ECOG PS 0-1. Para ser incluidos en el estudio, se requería que los pacientes presentaran niveles de bilirrubina total ≤ 1.5 mg/dL y aspartato aminotransferasa y alanina aminotransferasa (AST y ALT por sus siglas en inglés, respectivamente) ≤ 3 veces el valor del límite superior de la normalidad (upper limit of normal value - ULN, por sus siglas en inglés), o ≤ 5 veces ULN si existía metástasis hepática. Los pacientes fueron asignados de forma aleatoria en un ratio 2:1 para recibir una perfusión intravenosa de Cyramza® 8 mg/kg (n= 238) o placebo (n= 117) cada 2 semanas. La aleatorización se estratificó por pérdida de peso durante los 3 meses anteriores (≥ 10 % frente a <10 %), región geográfica y localización del tumor primario (gástrico frente GEJ). Las características basales demográficas y de la enfermedad estaban balanceadas. ECOG PS fue 1 para el 72 % de los pacientes. No se reclutaron en el estudio REGARD pacientes con cirrosis hepática Child-Pugh B o C. El 11 % de los pacientes tratados con Cyramza® y el 6 % de los pacientes tratados con placebo interrumpieron el tratamiento debido a las reacciones adversas. La supervivencia global mejoró de forma estadísticamente significativa en los pacientes que recibieron Cyramza® en comparación con aquellos que recibieron placebo (HR 0.776; IC 95 %: 0.603 a 0.998; p= 0.0473), correspondiendo a una reducción del riesgo de muerte del 22 % y a un aumento de la mediana de la supervivencia de 5.2 meses para Cyramza® y 3.8 meses para placebo. La supervivencia libre de progresión mejoró de forma estadísticamente significativa en pacientes que recibieron Cyramza® comparado con aquellos que recibieron placebo (HR 0.483; IC 95 %: 0.376 a 0.620; p < 0.0001), correspondiendo a una reducción del riesgo de progresión o muerte del 52 % y a un aumento en la mediana de PFS de 2.1 meses para Cyramza® y de 1.3 meses para placebo. En la Tabla 2 se muestran los resultados de eficacia.
Partiendo de los limitados datos disponibles del estudio REGARD sobre pacientes con cáncer gástrico HER2 positivo o adenocarcinoma de GEJ y sobre pacientes previamente tratados con trastuzumab (en RAINBOW), se considera poco probable que Cyramza® tenga un efecto perjudicial o que no tenga efecto en pacientes con cáncer gástrico HER2 positivo. Los análisis de subgrupos no estratificados realizados a posteriori de los pacientes del estudio RAINBOW previamente tratados con trastuzumab (n=39) sugirieron un beneficio en la supervivencia de pacientes (HR 0.679, IC 95 % 0.327, 1.419) y demostraron un beneficio en la PFS (HR 0.399, IC 95 % 0.194, 0.822). Cáncer colorrectal: RAISE: RAISE fue un ensayo global, aleatorizado, doble ciego de Cyramza® más FOLFIRI frente a placebo más FOLFIRI, en pacientes con mCRC, que tuvieron progresión de la enfermedad durante o tras la primera línea de tratamiento con bevacizumab, oxaliplatino y una fluoropirimidina. Los pacientes debían presentar ECOG PS 0 o 1 y progresión de la enfermedad en los 6 meses tras la última dosis del tratamiento de primera línea. Los pacientes debían presentar una función hepática, renal y una coagulación adecuadas. Se excluyeron a los pacientes con antecedentes hereditarios o adquiridos de sangrado no controlado o de trastornos trombóticos, antecedentes recientes de sangrado grave (Grado ≥ 3) o a aquellos que presentaban enfermedad tromboembólica arterial (ATE, por sus siglas en inglés) en los 12 meses anteriores a la aleatorización. También se excluyeron a aquellos pacientes que presentaron un ATE, hipertensión Grado 4, proteinuria Grado 3, acontecimiento hemorrágico Grado 3-4 o perforación intestinal durante el tratamiento en primera línea con bevacizumab. Un total de 1072 pacientes fueron aleatorizados (1:1) a recibir Cyramza® (n=536) a 8 mg/kg o placebo (n=536), en combinación con FOLFIRI. Todos los medicamentos se administraron de forma intravenosa. El régimen con FOLFIRI fue: Irinotecán 180 mg/m2 administrado durante 90 minutos y ácido folínico 400 mg/m2 administrado de forma simultánea durante 120 minutos, seguido de un bolo de 5-fluorouracilo (5-FU) 400 mg/m2 durante 2 a 4 minutos, seguido de 5-FU 2400 mg/m2 administrados por perfusión continua durante 46 a 48 horas. Los ciclos de tratamiento en ambos brazos se repitieron cada 2 semanas. A los pacientes que interrumpieron 1 o más componentes del tratamiento debido a una reacción adversa, se les permitió continuar el tratamiento con el resto de componente(s) hasta progresión de la enfermedad o toxicidad inaceptable. La variable primaria fue OS y la variable secundaria incluyó PFS, ORR y calidad de vida (QoL, por sus siglas en inglés) usando el criterio de la Organización Europea para la Investigación del Tratamiento del Cáncer (European Organisation for Research and Treatment of Cancer - EORTC, por sus siglas en inglés) QLQ-C30. La aleatorización se estratificó según región geográfica, mutación KRAS (presencia de mutación o no) y tiempo hasta progresión de la enfermedad (time to disease progression - TTP, por sus siglas en inglés) tras comienzo de la primera línea de tratamiento (< 6 meses frente a ≥ 6 meses).
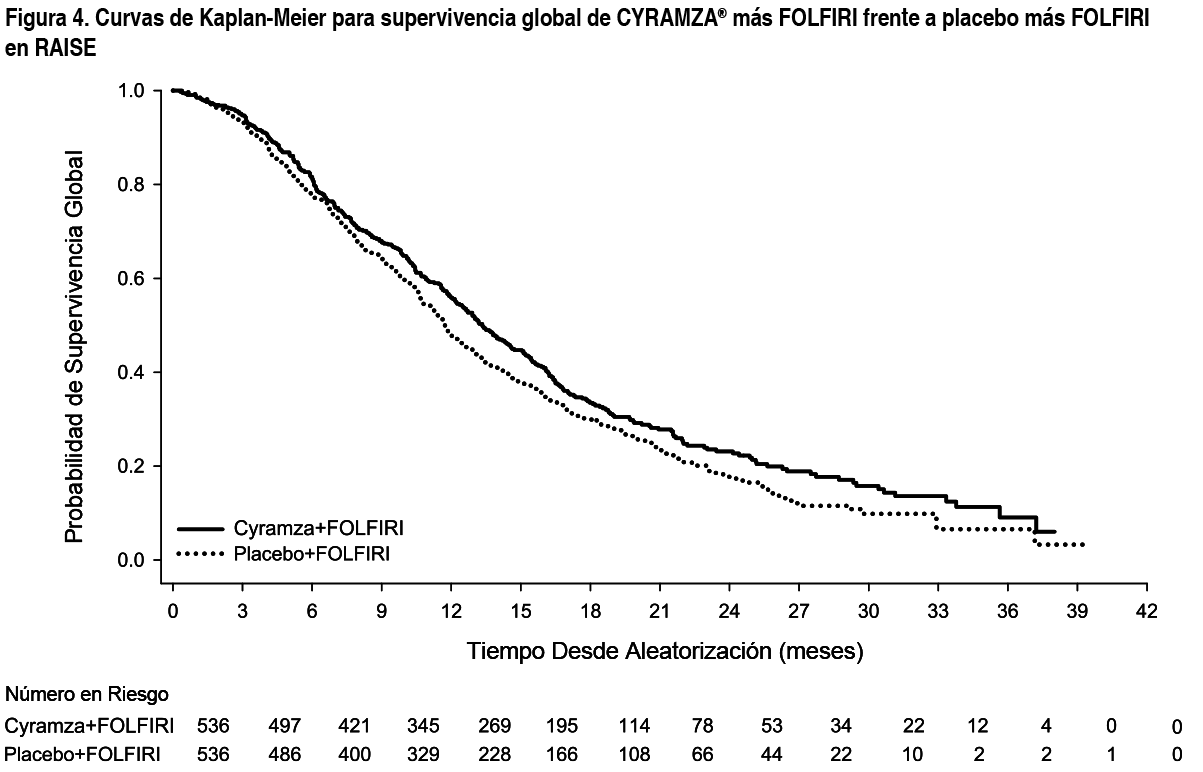
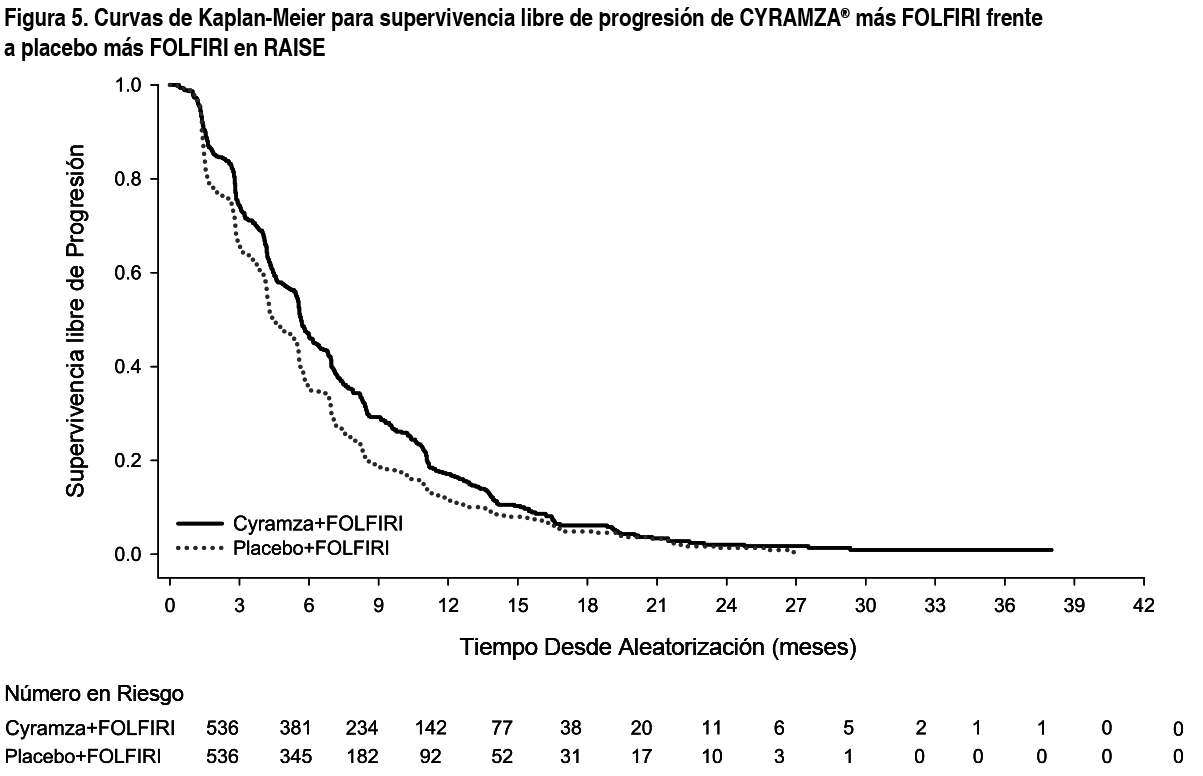
Las características demográficas y basales de la población ITT fueron similares en ambos brazos del tratamiento. La mediana de edad fue 62 ańos y el 40 % de pacientes tenían ≥ 65 ańos; 57 % de los pacientes eran hombres; 76 % eran blancos y el 20 % asiáticos; 49 % tuvieron ECOG PS 0; 49 % de los pacientes presentaron mutación KRAS en el tumor y 24 % de los pacientes tuvieron TTP < 6 meses tras el comienzo del tratamiento en primera línea. Se administró tratamiento anticanceroso sistémico posinterrupción al 54 % de los pacientes que recibieron Cyramza® más FOLFIRI y al 56 % de los pacientes que recibieron placebo más FOLFIRI. La supervivencia global mejoró de forma estadísticamente significativa en pacientes que recibieron Cyramza® más FOLFIRI comparada con aquellos que recibieron placebo más FOLFIRI (HR 0.844; IC 95 %: 0.730 a 0.976; p=0.0219). Hubo un aumento en la mediana de supervivencia de 1.6 meses a favor del brazo de Cyramza® más FOLFIRI: 13.3 meses en el brazo de Cyramza® más FOLFIRI y 11.7 meses en el brazo de placebo más FOLFIRI. La supervivencia libre de progresión mejoró de forma estadísticamente significativa en pacientes que recibieron Cyramza® más FOLFIRI comparado con aquellos que recibieron placebo más FOLFIRI (HR 0.793; IC 95 %: 0.697 a 0.903; p=0.0005). Hubo un aumento en la mediana de PFS de 1.2 meses a favor del brazo de Cyramza® más FOLFIRI: 5.7 meses en el brazo de Cyramza® más FOLFIRI y 4.5 meses en el brazo de placebo más FOLFIRI. Los resultados de eficacia se muestran en la Tabla 3 y en las Figuras 4 y 5. Se llevaron a cabo análisis preespecificados para OS y PFS según factores de estratificación. El HR de OS fue 0.82 (IC 95 %: 0.67 a 1.0) en pacientes con tumor KRAS no mutado y 0.89 (IC 95 %: 0.73 a 1.09) en pacientes con un tumor con la presencia de la mutación KRAS. Para pacientes con TTP ≥ 6 meses tras comenzar el tratamiento de primera línea, el HR de OS fue 0.86 (IC 95 %: 0.73 a 1.01) y en pacientes con TTP < 6 meses tras comenzar la primera línea de tratamiento, el HR fue 0.86 (IC 95 %: 0.64 a 1.13). Los análisis por subgrupos preespecificados tanto para PFS como OS según la edad (< 65 ańos y ≥ 65 ańos), sexo, raza, ECOG PS (0 o ≥ 1), número de órganos afectados, únicamente metástasis hepática, localización del tumor primario (colon o recto), niveles de antígenos carcinoembrionarios (< 200 μg/L, ≥ 200 μg/L), mostraron un efecto favorable del tratamiento con Cyramza® más FOLFIRI sobre placebo más FOLFIRI. En 32 de los 33 análisis por subgrupos preespecificados para OS, el HR fue < 1.0. El único subgrupo con HR > 1 fue el de pacientes con progresión de la enfermedad desde el comienzo del tratamiento en primera línea con bevacizumab < 3 meses (HR 1.02 [IC 95 %: 0.68 a 1.55]). Este único subgrupo puede considerarse que presenta una enfermedad agresiva relativamente refractaria al tratamiento en primera línea. En ambos grupos de tratamiento los pacientes que presentaron neutropenia tuvieron una mediana de OS más larga que la de aquellos que no presentaron neutropenia. La mediana de OS fue mayor en los pacientes con neutropenia de cualquier grado en el grupo de Cyramza® (16.1 meses) que en el grupo de placebo (12.6 meses). La mediana de OS en pacientes que no presentaron neutropenia fue 10.7 meses en ambos grupos.
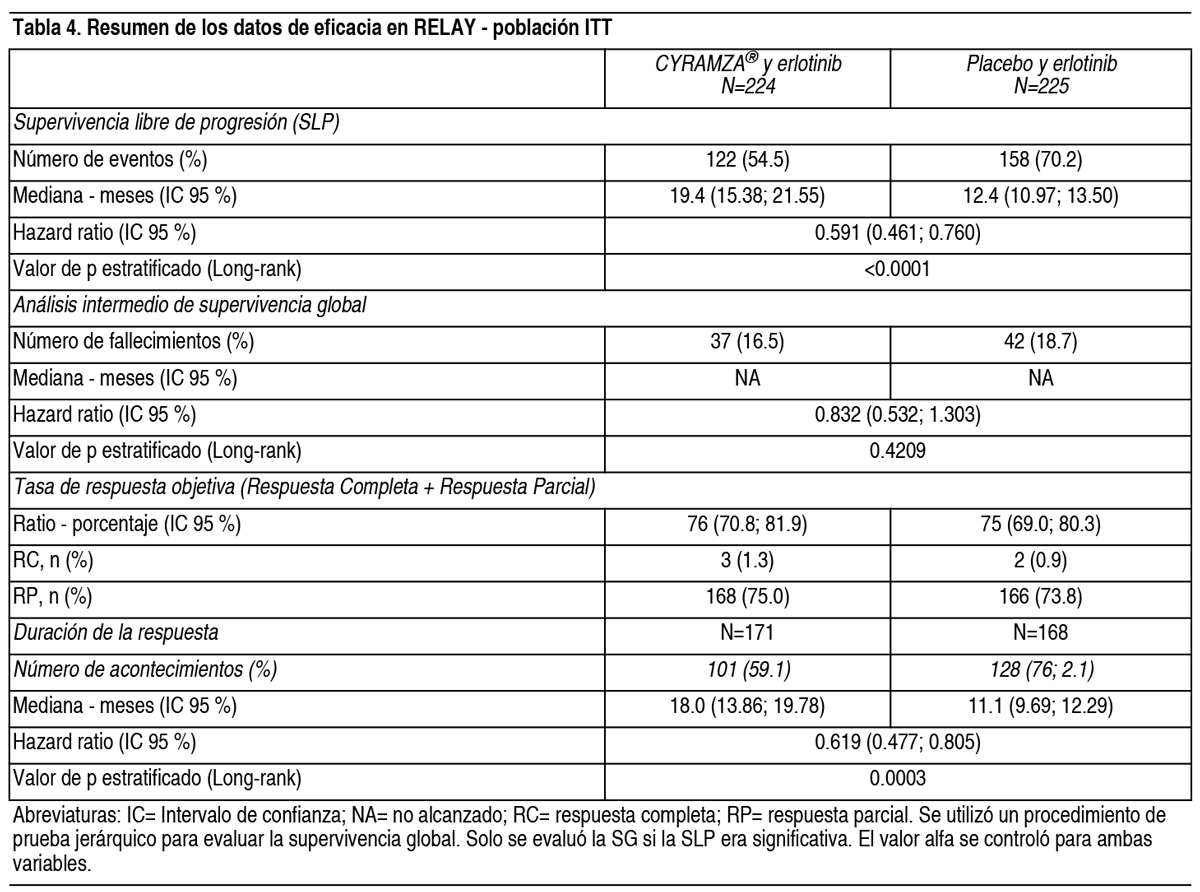
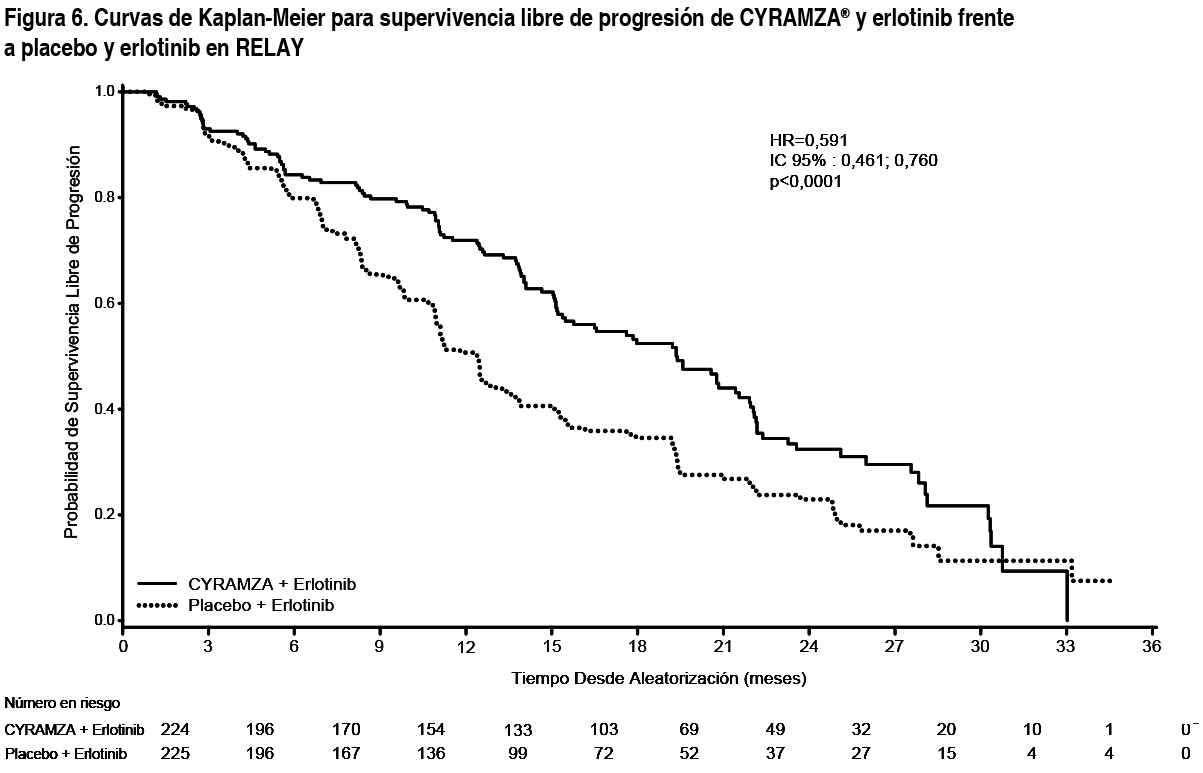
El ORR fue similar en ambos grupos de tratamiento (13.4 % frente a 12.5 %, Cyramza® más FOLFIRI frente a placebo más FOLFIRI, respectivamente). La tasa de control de la enfermedad (respuesta completa más respuesta parcial más enfermedad estable) fue numéricamente más alta en pacientes en el grupo de Cyramza® más FOLFIRI comparado con el grupo de placebo más FOLFIRI (74.1 % frente a 68.8 %, respectivamente). Para la EORTC QLQ-C30, los pacientes en el grupo de tratamiento de Cyramza® más FOLFIRI notificaron un descenso transitorio en QoL en la mayoría de las escalas comparado con los pacientes en el grupo de tratamiento de placebo más FOLFIRI. Las diferencias entre los grupos se notificaron tras el primer mes de tratamiento. Cáncer de Pulmón No Microcítico (CPNM): RELAY: RELAY fue un ensayo global, aleatorizado, doble ciego, fase 3 de Cyramza® y erlotinib frente a placebo y erlotinib que aleatorizó (1:1) 449 pacientes no tratados previamente con cáncer de pulmón no microcítico metastásico con mutaciones activadoras (deleción del exón 19 o exón 21 [L858R]) del receptor del factor de crecimiento epidérmico (EGFR, por sus siglas en inglés) al inicio del estudio. Los pacientes elegibles tenían ECOG PS 0 o 1. Se excluyeron del estudio los pacientes con metástasis en el SNC o presencia al inicio del estudio de las mutaciones activadoras del EGFR T790M. Se excluyeron también del estudio los pacientes con alto riesgo de sangrado, riesgo de eventos cardiovasculares, incluyendo aquellos que habían experimentado cualquier evento tromboembólico arterial en los 6 meses previos al reclutamiento. Las características demográficas y basales estaban equilibradas entre los brazos. El 77 % de pacientes eran asiáticos y el 22 % caucásicos. Los pacientes tratados con Cyramza® y erlotinib experimentaron una mejora estadística y clínicamente significativa en supervivencia libre de progresión (SLP) en comparación con los pacientes tratados con placebo y erlotinib (Tabla 4). Se observaron resultados consistentes entre los subgrupos que incluían deleciones del exón 19 y sustitución del exón 21 (L858R), edad, raza (caucásica HR: 0.618, asiática HR: 0.638), fumadores y no fumadores. Los datos de supervivencia global eran inmaduros en el momento del análisis final de SLP (17.6 % de madurez). Los resultados de eficacia del estudio RELAY se muestran en la Tabla 4 y Figura 6.
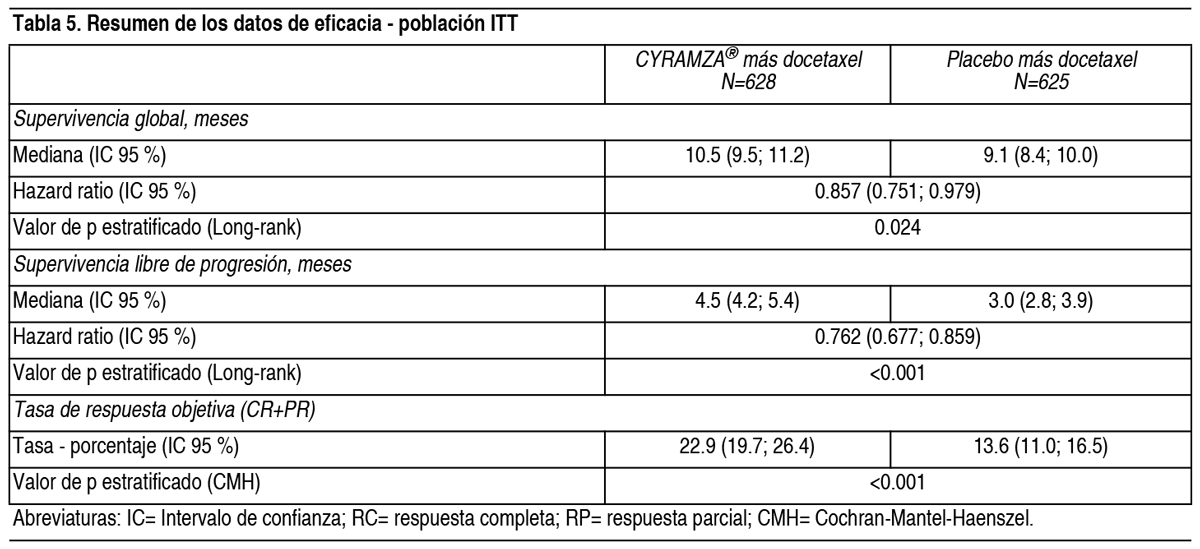
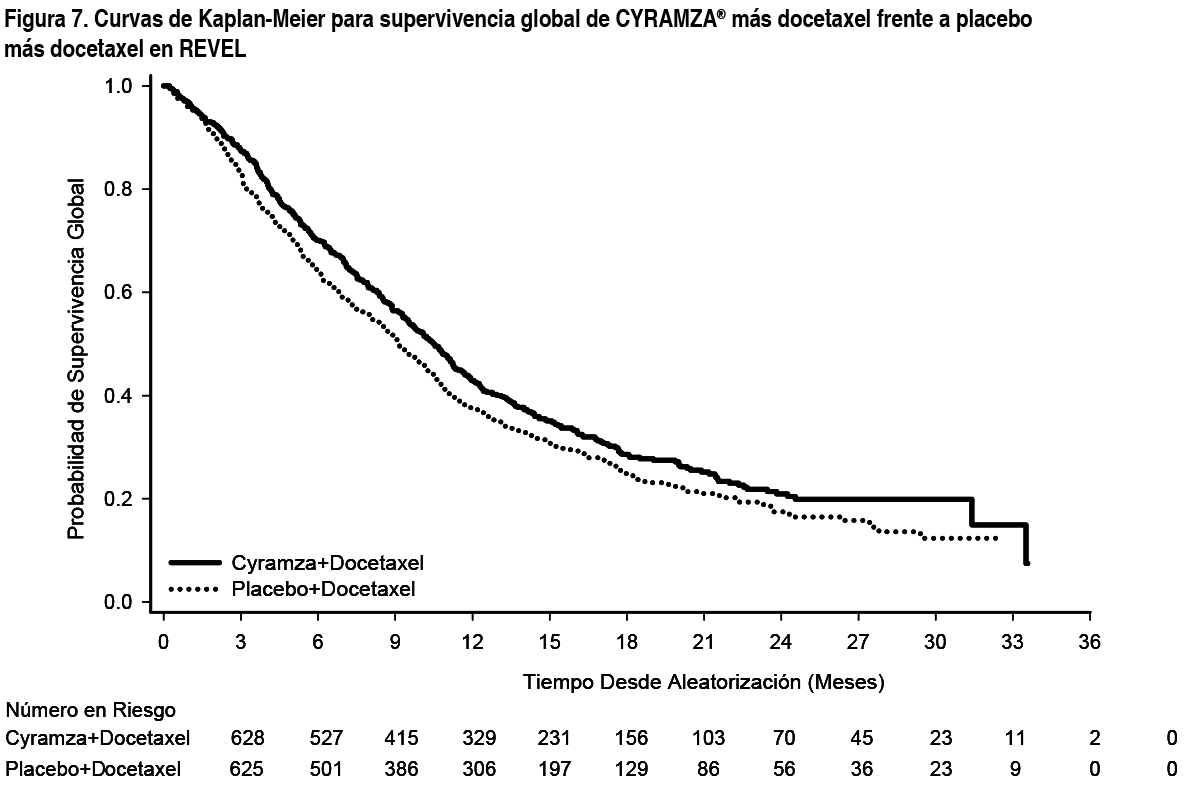
REVEL: REVEL es un ensayo aleatorizado, doble ciego de Cyramza® más docetaxel frente a placebo más docetaxel, llevado a cabo en 1253 pacientes con CPNM localmente avanzado o metastásico escamoso o no escamoso con progresión de la enfermedad durante o tras tratamiento basado en platino. La variable primaria fue OS. Los pacientes fueron aleatorizados en un ratio 1:1 para recibir Cyramza® más docetaxel (n=628) o placebo más docetaxel (n=625). La aleatorización fue estratificada por región geográfica, sexo, mantenimiento previo y ECOG PS. Cyramza® a 10 mg/kg o placebo y docetaxel a 75 mg/m˛ fueron cada uno administrados por perfusión intravenosa el día 1 de un ciclo de 21 días. Los hospitales en Asia Oriental administraron una dosis reducida de docetaxel a 60 mg/m˛ cada 21 días. Se excluyeron pacientes con hemorragia pulmonar o gastrointestinal grave y reciente, sangrado posquirúrgico, evidencia de hemorragias del SNC, compromiso tumoral de las vías respiratorias principales o vasos sanguíneos, cavitación intratumoral y antecedentes de sangrados significativos o trastornos trombóticos incontrolados. También se excluyeron pacientes que habían recibido cualquier tipo de terapia anticoagulante y/o terapia crónica con fármacos antiinflamatorios no esteroideos u otros agentes antiplaquetarios o aquellos con metástasis del SNC/cerebral clínicamente inestable no tratada. Se permitió el uso de aspirina a dosis de hasta 325 mg/día (ver sección Advertencias y Precauciones Especiales de Uso). Se incluyeron un número limitado de no caucásicos, especialmente pacientes negros (2.6 %). Por lo tanto, existe experiencia limitada con la combinación de ramucirumab y docetaxel en esos pacientes con CPNM avanzado, así como en pacientes con insuficiencia renal, enfermedad cardiovascular y obesidad. Las características demográficas basales de los pacientes y de la enfermedad estuvieron generalmente equilibradas entre los brazos: la mediana de edad fue 62 ańos; 67 % de los pacientes fueron hombres; 82 % fueron caucásicos, 13 % asiáticos; el ECOG PS fue 0 para el 32 % de los pacientes, 1 para el 67 % de los pacientes; el 73 % de los pacientes tuvieron histología no escamosa y el 26 % tuvieron histología escamosa. La terapia previa más frecuente fue pemetrexed (38 %), gemcitabina (25 %), taxano (24 %) y bevacizumab (14 %); 22 % de los pacientes recibieron terapia de mantenimiento previa. La mediana de la duración de la terapia de docetaxel fue 14.1 semanas para el brazo de Cyramza® más docetaxel (con una mediana de 4.0 perfusiones recibidas) y 12.0 semanas para el brazo de placebo más docetaxel (con una mediana de perfusiones recibidas de 4.0). La OS aumentó de forma estadísticamente significativa en pacientes que recibieron Cyramza® más docetaxel comparado con aquellos que recibieron placebo más docetaxel (HR 0.857; IC 95 %: 0.751 a 0.979; p=0.024). Hubo un aumento en la mediana de supervivencia de 1.4 meses a favor del brazo de Cyramza® más docetaxel: 10.5 meses en el brazo de Cyramza® más docetaxel y 9.1 meses en el brazo de placebo más docetaxel. PFS aumentó de forma estadísticamente significativa en pacientes que recibieron Cyramza® más docetaxel comparado con aquellos que recibieron placebo más docetaxel (HR 0.762; IC 95 %: 0.677 a 0.859; p<0.001). Hubo un aumento en la mediana de PFS de 1.5 meses a favor del brazo de Cyramza® más docetaxel: 4.5 meses en el brazo de Cyramza® más docetaxel y 3 meses en el brazo de placebo más docetaxel. ORR mejoró de forma significativa en pacientes que recibieron Cyramza® más docetaxel comparada con aquellos que recibieron placebo más docetaxel (22.9 % vs. 13.6 %, p<0.001). El análisis primario de QoL mostró un tiempo hasta el deterioro similar entre los brazos de tratamiento según todas las puntuaciones de las Escalas de Síntomas de Cáncer de Pulmón (Lung Cancer Symptom Scale - LCSS, por sus siglas en inglés). Se observó una mejora invariable (Cyramza® más docetaxel vs. placebo más docetaxel) en subgrupos importantes para PFS y OS. Los resultados de los subgrupos para OS mostraron: histología no escamosa [HR 0.83; IC 95 %: 0.71 a 0.97; mediana de OS (mOS): 11.1 vs. 9.7 meses] e histología escamosa (HR 0.88; IC 95 %: 0.069 a 1.13; mOS: 9.5 vs. 8.2 meses); pacientes con mantenimiento previo (HR 0.69; IC 95 %: 0.51 a 0.93; mOS: 14.4 vs. 10.4 meses); tiempo desde el comienzo de la terapia previa < 9 meses (HR 0.75; IC 95 %: 0.64 a 0.88; mOS: 9.3 vs. 7.0 meses); pacientes < 65 ańos (HR 0.74, IC 95 %: 0.62, 0.87; mOS: 11.3 vs. 8.9 meses). Se ha observado una tendencia a menor eficacia conforme aumenta la edad del paciente que recibe Cyramza® más docetaxel para el tratamiento de CPNM avanzado con progresión de la enfermedad tras quimioterapia basada en platino (ver sección Propiedades Farmacodinámicas). No se observaron diferencias de eficacia entre los grupos de tratamiento en los subgrupos de pacientes ≥ 65 ańos (OS HR 1.10, IC 95 %: 0.89, 1.36; mOS: 9.2 vs. 9.3 meses, ver sección Advertencias y Precauciones Especiales de Uso), pacientes tratados previamente con taxanos (HR 0.81; IC 95 %: 0.62 a 1.07; mOS 10.8 vs. 10.4 meses) y aquellos con tiempo desde comienzo de la terapia previa ≥ 9 meses (HR 0.95; IC 95 %: 0.75 a 1.2; mOS: 13.7 vs. 13.3 meses). Los resultados de eficacia se muestran en la Tabla 5.
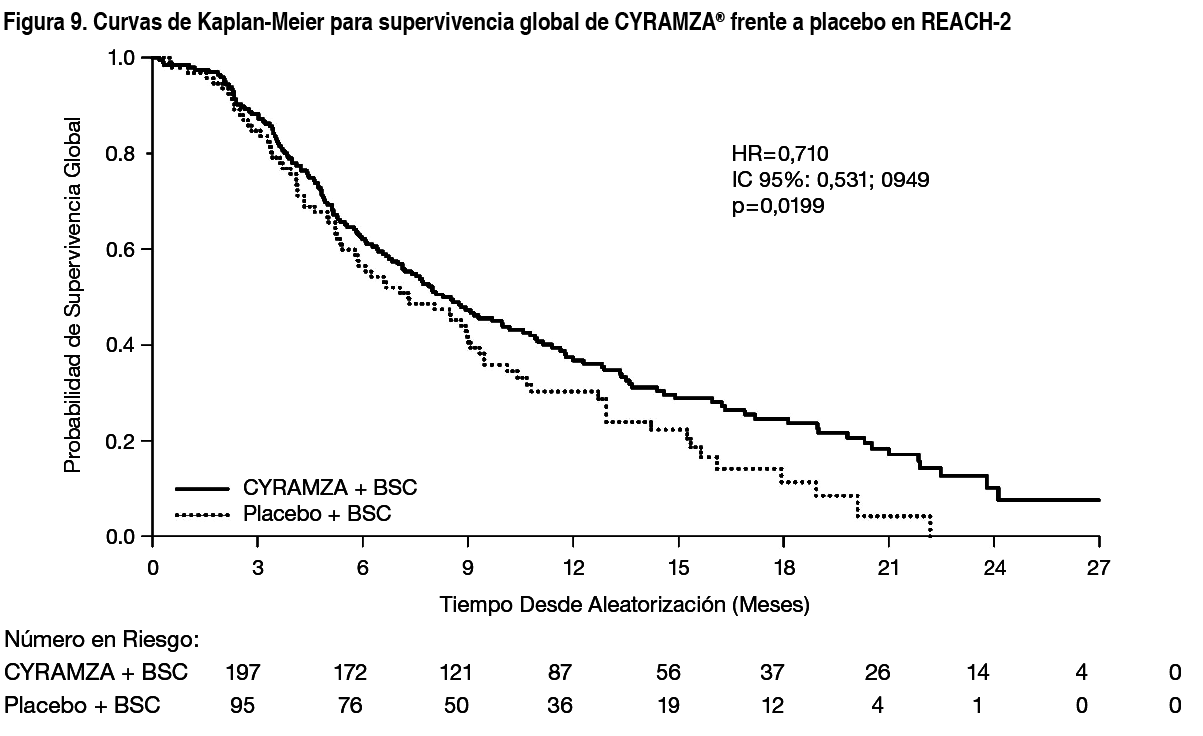
Carcinoma Hepatocelular (CHC): REACH-2: REACH-2 fue un estudio global, aleatorizado, doble ciego de Cyramza® con BSC frente a placebo más BSC que aleatorizó (2:1) a 292 pacientes con CHC que tenían una alfafetoproteína (AFP) sérica ≥ 400 ng/ml a la entrada en el estudio. Los pacientes incluidos en el estudio tuvieron una progresión de la enfermedad durante o después del tratamiento previo con sorafenib o eran intolerantes a sorafenib. Los pacientes elegidos eran Child-Pugh A (puntuación < 7), tenían aclaramiento de creatinina ≥ 60 ml/min y ECOG PS de 0 o 1. Además, los pacientes tenían tanto cáncer de hígado estadio B de Barcelona Clinic Liver Cancer (BCLC, por sus siglas en inglés) y ya no eran susceptibles de tratamiento locorregional, como estadio C de BCLC. Fueron excluidos del estudio pacientes con metástasis cerebrales, enfermedad leptomeníngea, compresión descontrolada de la médula espinal, antecedentes de encefalopatía hepática o en la actualidad, o ascitis clínicamente significativa, hemorragia grave por várices en los 3 meses previos al tratamiento, o várices gástricas o esofágicas con un riesgo alto de sangrado. La variable principal fue la supervivencia global. Como criterio de inclusión en el estudio REACH-2 se estableció el umbral de la AFP elevada obtenido en base a los resultados de supervivencia en un análisis exploratorio de un subgrupo preespecificado del estudio REACH (un estudio clínico fase 3 finalizado previamente en 565 pacientes con CHC aleatorizado (1:1) a Cyramza® más BSC o a placebo más BSC que tuvieron progresión de la enfermedad durante o después del tratamiento previo con sorafenib). En el estudio REACH-2, las características demográficas de los pacientes y las características de la enfermedad de inicio estaban bien equilibradas en ambos brazos, a excepción de la AFP, que fue inferior en el brazo de placebo. Los pacientes tratados con Cyramza® experimentaron un incremento estadísticamente significativo en la SG, en comparación con placebo (Tabla 6). El principal resultado de eficacia en el estudio REACH-2 fue apoyado por un incremento estadísticamente significativo en la supervivencia libre de progresión en pacientes tratados con Cyramza® en comparación con pacientes tratados con placebo. El efecto relativo del tratamiento (evaluado por HR) de Cyramza® en comparación con placebo fue generalmente consistente en todos los subgrupos, incluidos la edad, la raza, la etiología de la enfermedad y el motivo de la interrupción del tratamiento con sorafenib (progresión de la enfermedad frente a intolerancia). En el estudio REACH-2 se observó una asociación relevante entre la exposición y la eficacia para ramucirumab (ver sección Propiedades Farmacocinéticas). Los resultados de eficacia del estudio REACH-2 se muestran en la Tabla 6 y la Figura 9.
Pacientes con estado funcional (PS) ≥ 2 según la escala Eastern Cooperative Oncology Group (ECOG): Los pacientes con un ECOG PS ≥ 2 fueron excluidos de los ensayos pivotales en todas las indicaciones, por lo que se desconoce la seguridad y eficacia de Cyramza® en este grupo de pacientes. Inmunogenicidad: Los pacientes en 2 estudios fase 3, RAINBOW y REGARD, fueron examinados en numerosos momentos para comprobar los anticuerpos antifármaco (anti-drug antibodies - ADA, por sus siglas en inglés). Se evaluaron muestras de 956 pacientes: 527 pacientes tratados con Cyramza® y 429 pacientes del grupo control. 11 (2.2 %) de los pacientes tratados con Cyramza® y 2 (0.5 %) de los pacientes control desarrollaron ADA. Ninguno de los pacientes con ADA presentó reacciones relacionadas con la perfusión (infusion related reactions - IRR, por sus siglas en inglés). Ningún paciente desarrolló anticuerpos neutralizadores de ramucirumab. No hay datos suficientes para evaluar los efectos de ADA sobre la eficacia o seguridad de ramucirumab. Población pediátrica: La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con Cyramza® en todos los grupos de la población pediátrica en adenocarcinoma gástrico, adenocarcinoma de colon y recto, en carcinoma de pulmón, y cáncer de hígado (ver sección Posología y Modo de Administración para consultar la información sobre el uso en la población pediátrica). Propiedades Farmacocinéticas: Tras la pauta posológica de 8 mg/kg cada 2 semanas, la media geométrica de la concentración mínima de ramucirumab (Cmín) en suero en pacientes con cáncer gástrico avanzado antes de la administración de la cuarta y séptima dosis de ramucirumab como agente único fue 49.5 μg/ml (rango de 6.3-228 μg/ml) y 74.4 μg/ml (rango 13.8-234 μg/ml), respectivamente. La media geométrica de la Cmín en suero de pacientes con CHC de ramucirumab tras la administración de la segunda, cuarta y séptima dosis de ramucirumab fueron 23.5 μg/ml (rango de 2.9-76.5 μg/ml), 44.1 μg/ml (rango de 4.2-137 μg/ml) y 60.2 μg/ml (rango de 18.3-123 μg/ml), respectivamente. Tras la pauta posológica de 8 mg/kg cada 2 semanas de ramucirumab en combinación con FOLFIRI, las medias geométricas de la Cmínen el suero de pacientes con mCRC de ramucirumab fueron 46.3 μg/ml (rango de 7.7-119 μg/ml) y 65.1 μg/ml (rango de 14.5-205 μg/ml) antes de la administración de la tercera y quinta dosis respectivamente. Tras la pauta posológica de 10 mg/kg cada 3 semanas de ramucirumab, las medias geométricas de la Cmínmín de ramucirumab en el suero de pacientes con CPNM fueron 28.3 μg/ml (rango de 2.5-108 μg/ml) y 38.4 μg/ml (rango de 3.1-128 μg/ml) antes de la administración de la tercera y quinta dosis, respectivamente de ramucirumab administrado en combinación con docetaxel. Tras la pauta posológica de 10 mg/kg cada 2 semanas de ramucirumab, las medias geométricas de la Cmín de ramucirumab en el suero de pacientes con CPNM fueron 68.5 μg/ml (rango de 20.3-142 μg/ml) y 85.7 μg/ml (rango de 36.0-197 μg/ml) antes de la administración de la cuarta y séptima dosis, respectivamente de ramucirumab administrado en combinación con erlotinib. Absorción: Cyramza® se administra por perfusión intravenosa. No se han llevado a cabo estudios con otras vías de administración. Distribución: El volumen de distribución medio en el estado estacionario fue de 5.4 l (15 %) (% coeficiente de variación [CV%]) basado en la aproximación por un modelo farmacocinético poblacional (population pharmacokinetic approach - PopPK, por sus siglas en inglés). Biotransformación: No se ha estudiado el metabolismo de ramucirumab. El aclaramiento de los anticuerpos es principalmente por catabolismo. Eliminación: El aclaramiento medio (CV%) de ramucirumab fue 0.015 L/hora (30 %) y el valor medio de vida media fue 14 días (20 %), según PopPK. Dependencia de tiempo y dosis: No hubo una desviación clara de la proporcionalidad de dosis en la farmacocinética de ramucirumab desde 6 mg/kg a 20 mg/kg. Se observó un ratio acumulado de 1.5 para ramucirumab cuando se administró una dosis cada 2 semanas. Según simulaciones usando el modelo PopPK, el estado estacionario debería alcanzarse en la sexta dosis. Pacientes de edad avanzada: No hubo diferencias en la exposición a ramucirumab en pacientes ≥ 65 ańos comparado con pacientes < 65 ańos, según el modelo PopPK. Insuficiencia renal: No se han llevado a cabo estudios específicos para evaluar el efecto de la insuficiencia renal sobre la farmacocinética de ramucirumab. Según modelos PopPK, la exposición a ramucirumab fue parecida en pacientes con insuficiencia renal leve (aclaramiento de creatinina (creatinine clearance - CrCl, por sus siglas en inglés) ≥ 60 a < 90 ml/min), moderada (CrCl ≥ 30 a < 60 ml/min) o grave (CrCl 15 a 29 ml/min) comparado con pacientes con función renal normal (CrCl ≥ 90 ml/min). Insuficiencia hepática: No se han llevado a cabo estudios específicos para evaluar los efectos de la insuficiencia hepática sobre la farmacocinética de ramucirumab. Según modelos PopPK, la exposición a ramucirumab en pacientes con insuficiencia hepática leve (niveles de bilirrubina total >1.0-1.5 veces el valor de ULN y cualquier valor de AST o bilirrubina total de ≤ 1.0 veces el valor de ULN y AST > ULN) o con insuficiencia hepática moderada (niveles de bilirrubina total > 1.5-3.0 veces el valor de ULN y cualquier valor de AST) fue similar a la de pacientes con función hepática normal (niveles de bilirrubina total y AST ≤ a ULN). Ramucirumab no se ha estudiado en pacientes con insuficiencia hepática grave (niveles de bilirrubina total > 3.0 veces ULN y cualquier valor de AST). Otras poblaciones especiales: Según modelos PopPK, se encontró que las siguientes covariables no presentaron impacto sobre el comportamiento farmacocinético de ramucirumab: edad, sexo, raza, y niveles de albúmina. Estos y otros factores investigados tuvieron < 20 % de efecto en el comportamiento farmacocinético de ramucirumab. El peso corporal se considera una covariable significativa de la farmacocinética de ramucirumab apoyando la dosificación basada en el peso corporal. Relación exposición-respuesta: Eficacia: Los análisis de la exposición-respuesta a través de los estudios pivotales, mostraron que la eficacia estaba relacionada con la exposición a ramucirumab. La eficacia, medida por incrementos en SG, se asoció con el aumento del rango de exposición a ramucirumab al administrarse a una dosis de 8 mg/kg cada 2 semanas y de 10 mg/kg de ramucirumab cada 3 semanas. Un incremento de la SLP también se asoció con el aumento de la exposición a ramucirumab para el cáncer gástrico avanzado, CPNM con progresión de la enfermedad tras quimioterapia basada en platino y mCRC. En el estudio REACH-2 para CHC, se observó una asociación relevante entre la exposición y la eficacia para ramucirumab, que mostró que solo los pacientes con una exposición superior a la mediana experimentaron un incremento en la SG, en comparación con placebo, y esta relación entre la exposición y la eficacia permanece constante después de tratar de ajustar la eficacia por otros factores pronósticos. Se observó un efecto del tratamiento sobre la PFS para todos los niveles de exposición al administrarse una dosis de 8 mg/kg de ramucirumab cada 2 semanas. No se observó esta relación en el estudio RELAY para CPNM con 10 mg/kg de ramucirumab y erlotinib administrado cada 2 semanas. Seguridad: En el estudio RAINBOW, la incidencia de hipertensión, neutropenia y leucopenia de Grado ≥ 3, aumentó con una mayor exposición a ramucirumab. En el estudio RAISE, la incidencia de neutropenia Grado ≥ 3 aumentó con una mayor exposición a ramucirumab. En el estudio RELAY, no se identificó una relación entre el perfil de seguridad y la exposición al fármaco para las variables seleccionadas de seguridad, incluyendo hipertensión Grado ≥ 3, diarrea, proteinuria y dermatitis acneiforme. En el estudio REVEL, la incidencia de neutropenia febril e hipertensión Grado ≥ 3 aumentó con una mayor exposición a ramucirumab. En los datos agrupados de los estudios REACH-2 y REACH (pacientes con alfafetoproteína ≥ 400 ng/ml), la incidencia de hipertensión Grado ≥ 3 aumentó con una mayor exposición a ramucirumab. Datos preclínicos sobre seguridad: No se han llevado a cabo estudios en animales para evaluar el potencial carcinogénico o genotoxicidad de ramucirumab. Los órganos identificados como diana en estudios de toxicidad a dosis repetidas en monos cynomolgus fueron rińón (glomerulonefritis), hueso (engrosamiento y osificación endocondral anormal de la placa de crecimiento epifisaria) y órganos reproductivos femeninos (descenso del peso de ovarios y útero). Se observó un grado mínimo de inflamación y/o infiltración de células mononucleares en varios órganos. No se llevaron a cabo estudios de toxicidad reproductiva con ramucirumab, sin embargo, existen modelos animales que asocian la angiogénesis, el VEGF y el Receptor 2 de VEGF a aspectos críticos de la reproducción en mujeres, el desarrollo embriofetal y el desarrollo postnatal. Basándose en el mecanismo de acción de ramucirumab, es probable que, en animales, ramucirumab pueda inhibir la angiogénesis y derivar en reacciones adversas sobre la fertilidad (ovulación), desarrollo de la placenta, desarrollo del feto y desarrollo postnatal. Una dosis única de ramucirumab no afectó a la curación de heridas en monos usando un modelo incisional de espesor completo.
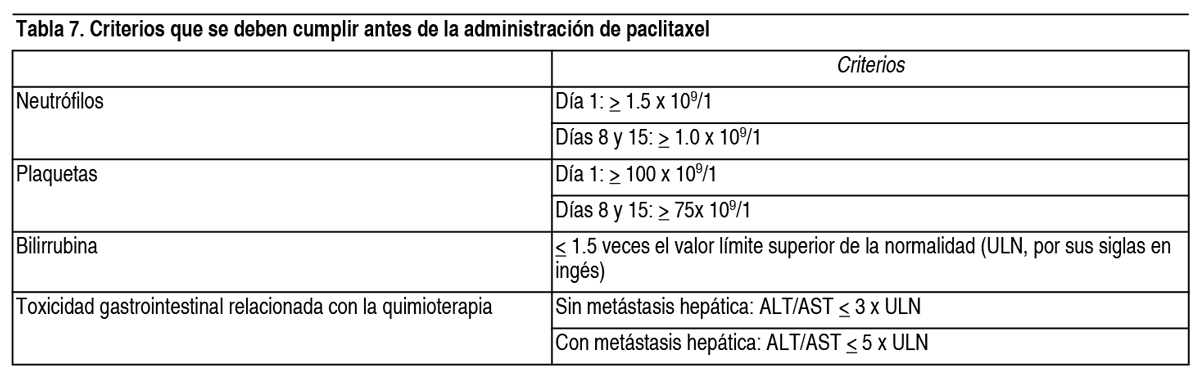
Posología: El tratamiento con Cyramza® se debe iniciar y estar bajo la supervisión de médicos con experiencia en oncología. Cáncer gástrico y adenocarcinoma de la unión gastroesofágica (GEJ por sus siglas en inglés): Cyramza® en combinación con paclitaxel: La dosis recomendada de Cyramza® es de 8 mg/kg los días 1 y 15 de un ciclo de 28 días, antes de la perfusión de paclitaxel. La dosis recomendada de paclitaxel es de 80 mg/m2 administrado por perfusión intravenosa durante aproximadamente 60 minutos los días 1, 8 y 15 de un ciclo de 28 días. Antes de cada perfusión de paclitaxel se debe realizar un hemograma completo y bioquímica sanguínea al paciente para evaluar la función hepática. En la tabla 7 se muestran los criterios que se deben cumplir antes de cada perfusión con paclitaxel.
Cyramza® como agente único: La dosis recomendada de Cyramza® como agente único es de 8 mg/kg cada 2 semanas. Cáncer colorrectal: La dosis recomendada de Cyramza® es 8 mg/kg cada 2 semanas administrada por percusión intravenosa antes de la administración de FOLFIRI. Antes de la quimioterapia, se debe realizar un hemograma completo al paciente. En la Tabla 8 se muestran los criterios que se deben cumplir antes de la administracion de FOLFIRI.
Cáncer de pulmón no microcítico (CPNM): Cyramza® en combinación con erlotinib para el tratamiento del CPNM con mutaciones activadoras de EGFR: La dosis recomendada de Cyramza® en combinación con erlotinib es 10 mg/kg cada 2 semanas. Antes del inicio del tratamiento con Cyramza® y erlotinib, se debe determinar la presencia de las mutaciones del EGFR utilizando un método de determinación validado. Consultar el prospecto (inserto) de erlotinib sobre posología y forma de administración de erlotinib. Cyramza® en combinación con docetaxel para el tratamiento del CPNM tras quimioterapia basada en platino: La dosis recomendada de Cyramza® es 10 mg/kg el día 1 de un ciclo de 21 días, antes de la perfusión de docetaxel. La dosis recomendada de docetaxel es 75 mg/m˛ administrada por perfusión intravenosa durante aproximadamente 60 minutos el día 1 de un ciclo de 21 días. Para pacientes de Asia Oriental, se debe considerar empezar con una dosis reducida de docetaxel de 60 mg/m2 el día 1 de un ciclo de 21 días. Consultar el prospecto (inserto) de docetaxel para consejos de dosificación específicos. Carcinoma hepatocelular (CHC): La dosis recomendada de Cyramza® como agente único es 8 mg/kg cada 2 semanas. Prueba de AFP en CHC: Se deben seleccionar pacientes con CHC en base a una concentración de AFP sérica ≥ 400 ng/ml determinada por un test de AFP validado previo al tratamiento con Cyramza® (ver sección Propiedades Farmacodinámicas). Duración del tratamiento: Se recomienda que el tratamiento continúe hasta progresión de la enfermedad o hasta toxicidad inaceptable. Medicación previa: Antes de la perfusión de Cyramza® se recomienda administrar al paciente un antagonista de histamina H1 (p. ej., difenhidramina) como medicación previa. Si un paciente presenta reacciones relacionadas con la perfusión de Grado 1 o 2, debe recibir medicación previa en todas las perfusiones posteriores. Se debe administrar dexametasona (o equivalente) si un paciente presenta una segunda reacción relacionada con la perfusión (IRR, por sus siglas en inglés) de Grado 1 o 2; y luego, para las siguientes perfusiones se deben utilizar como medicación previa los siguientes medicamentos o equivalentes: un antagonista de histamina H1 intravenoso (por ejemplo, difenhidramina hidrocloruro), paracetamol y dexametasona. Ver el prospecto (inserto) de paclitaxel, de los componentes de FOLFIRI y de docetaxel según proceda, para sus requerimientos sobre medicación previa e información adicional. Ajuste de dosis para Cyramza®: Reacciones relacionadas con la perfusión: si el paciente experimenta una IRR de Grado 1 o 2, la velocidad de perfusión de Cyramza® se debe reducir en un 50 %, así como en todas las perfusiones posteriores. En caso de IRR de Grado 3 o 4, se debe interrumpir el tratamiento con Cyramza® de forma inmediata y permanente (ver sección Advertencias y Precauciones Especiales de Uso). Hipertensión: Antes de cada administración de Cyramza® se debe controlar y tratar la tensión arterial según indicación clínica correspondiente. En caso de hipertensión grave, el tratamiento con Cyramza® se debe interrumpir temporalmente hasta que se controle con medicación. En caso de hipertensión médicamente significativa que no pueda ser controlada de forma segura con antihipertensivos, el tratamiento con Cyramza® se debe interrumpir de forma permanente (ver sección Advertencias y Precauciones Especiales de Uso). Proteinuria: Durante el tratamiento con Cyramza® se debe controlar el desarrollo o empeoramiento de la proteinuria en el paciente. Si los niveles de proteína en orina son ≥ 2+ en una tira reactiva de orina, se debe recoger una muestra de orina de 24 horas. El tratamiento con Cyramza® debe interrumpirse temporalmente en caso de niveles de proteína en orina ≥ 2 g/24 horas. Una vez que los niveles de proteínas en orina se hayan restablecido a valores < 2 g/24 horas, se debe reiniciar el tratamiento con una dosis reducida (ver Tabla 9). Si reaparecen niveles de proteína en orina ≥ 2 g/24 horas, se recomienda una segunda reducción de la dosis (ver Tabla 9). Si el nivel de proteinuria es > 3 g/24 horas o existe síndrome nefrótico, el tratamiento con ramucirumab se debe interrumpir de forma permanente.
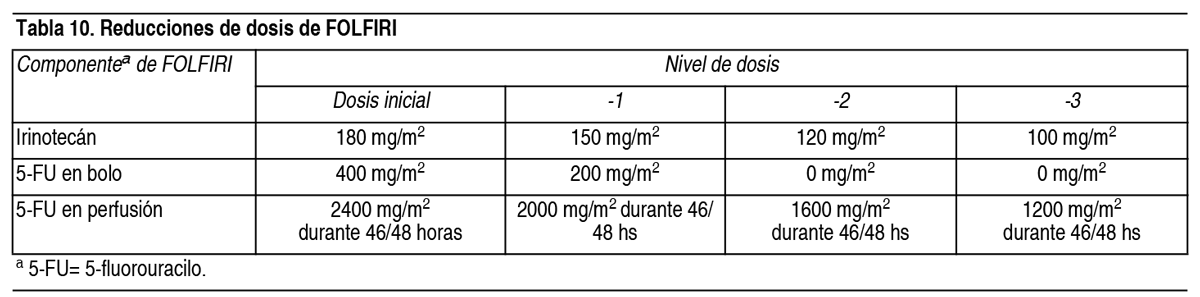
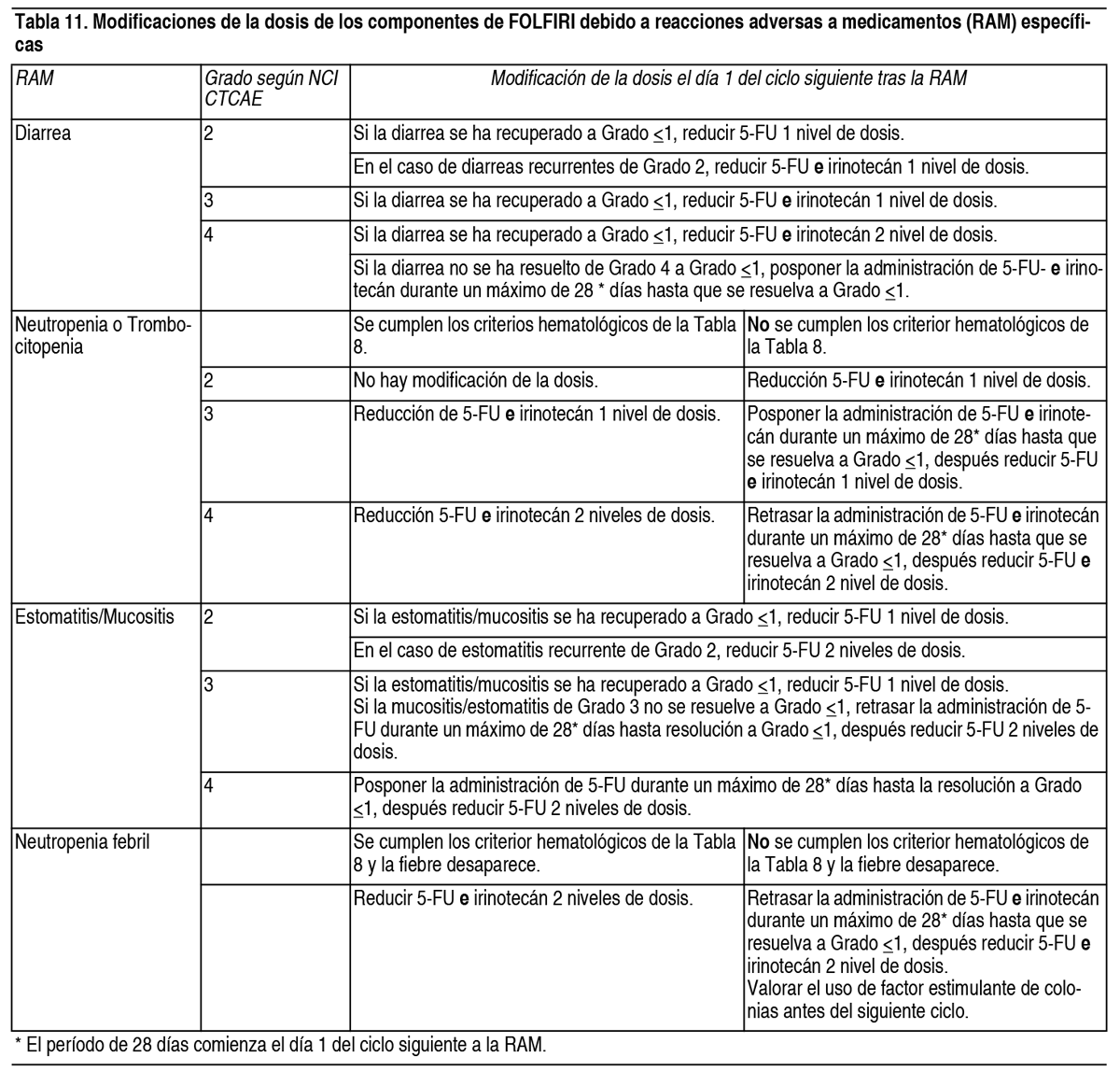
Cirugía programada o dificultad en la curación de heridas: Se debe interrumpir el tratamiento con Cyramza® temporalmente durante al menos 4 semanas previas a una cirugia programada. Si existen complicaciones en la curación de las heridas, el tratamiento con Cyramza® se debe interrumpir temporalmente hasta que la herida este completamente curada (ver sección Advertencias y Precauciones Especiales de Uso). Interrupción permanente: El tratamiento con Cyramza® se debe interrumpir permanentemente en los casos de: Enfermedad tromboembólica arterial grave (ver sección Advertencias y Precauciones Especiales de Uso). Perforaciones gastrointestinales (ver sección Advertencias y Precauciones Especiales de Uso). Hemorragias graves: hemorragias Grado 3 o 4 según NCI CTCAE (ver sección Advertencias y Precauciones Especiales de Uso). Desarrollo espontáneo de fístulas (ver sección Advertencias y Precauciones Especiales de Uso). Encefalopatía hepática o síndrome hepatorrenal (ver sección Advertencias y Precauciones Especiales de Uso). Ajuste de dosis de paclitaxel: Se deben aplicar reducciones de dosis de paclitaxel en función del grado de toxicidad del paciente. Para toxicidad hematológica Grado 4 según NCI CTCAE o toxicidad no hematológica relacionada con paclitaxel de Grado 3, se recomienda reducir la dosis de paclitaxel en 10 mg/m2 para todos los ciclos posteriores. Si la toxicidad persiste o es recurrente, se recomienda realizar una segunda reducción adicional de la dosis de 10 mg/m2. Ajuste de dosis de FOLFIRI: En el caso de toxicidades específicas, pueden aplicarse reducciones de dosis de los componentes individuales de FOLFIRI. Las modificaciones de las dosis de cada componente de FOLFIRI se deben aplicar de forma independiente y se muestran en la Tabla 10. La Tabla 11 muestra más información sobre retrasos de la administración o reducciones de dosis de los componentes de FOLFIRI para el siguiente ciclo basándose en el grado máximo de las reacciones adversas a medicamentos específicas.
Ajuste de dosis de docetaxel: Las reducciones de dosis de docetaxel se pueden aplicar basándose en el grado de toxicidad que ha presentado el paciente. El tratamiento con docetaxel se debe posponer hasta que la toxicidad se haya resuelto, en aquellos pacientes que durante el tratamiento presenten neutropenia febril, neutrófilos < 500 células/mm3 durante más de 1 semana, reacciones cutáneas graves o acumulativas u otras toxicidades Grado 3 o 4 no hematológicas. Se recomienda reducir la dosis de docetaxel en 10 mg/m2 para el resto de los ciclos. Se recomienda una segunda reducción en 15 mg/m2 si las toxicidades persisten o son recurrentes. En este caso, los pacientes de Asia Oriental con una dosis de inicio de 60 mg/m˛ deben interrumpir el tratamiento con docetaxel (ver sección Posología-Posología y Modo de Administración). Poblaciones especiales: Pacientes de edad avanzada: Los ensayos pivotales no han mostrado suficiente evidencia de que los pacientes de 65 ańos o mayores tengan un mayor riesgo de sufrir reacciones adversas comparando con los pacientes menores de 65 ańos. No se recomienda reducción de la dosis (ver secciones Advertencias y Precauciones Especiales de Uso y Propiedades Farmacodinámicas). Insuficiencia renal: No se han llevado a cabo estudios específicos con Cyramza® en pacientes con insuficiencia renal. Los datos clínicos sugieren que no se necesita un ajuste de dosis en pacientes con insuficiencia renal leve, moderada o insuficiencia renal grave (ver secciones Advertencias y Precauciones Especiales de Uso y Propiedades Farmacocinéticas). No se recomienda reducir la dosis. Insuficiencia hepática: No se han llevado a cabo estudios específicos con Cyramza® en pacientes con insuficiencia hepática. Los datos clínicos indican que no son necesarios ajustes de dosis en pacientes con insuficiencia hepática leve o moderada. No existen datos sobre la administración de Cyramza® en pacientes con insuficiencia hepática grave (ver secciones Advertencias y Precauciones Especiales de Uso y Propiedades Farmacocinéticas). No se recomienda reducir la dosis. Población pediátrica: No se ha establecido la seguridad y eficacia de Cyramza® en nińos y adolescentes (< 18 ańos). No existe una recomendación de uso específica para Cyramza® en la población pediátrica para la indicación de cáncer gástrico avanzado o adenocarcinoma gastroesofágico, adenocarcinoma de colon y recto, carcinoma de pulmón, y carcinoma hepatocelular. Forma de administración: Cyramza® se administra por vía intravenosa. Tras dilución, Cyramza® se administra en perfusión intravenosa durante aproximadamente 60 minutos. No se debe administrar en bolo intravenoso o inyección rápida. Para alcanzar la duración de perfusión requerida de 60 minutos aproximadamente, la velocidad de perfusión máxima no debe exceder los 25 mg/minuto; en caso contrario la duración de la perfusión debe ser incrementada. Durante la perfusión, los pacientes deben ser monitorizados ante la posible aparición de signos de reacciones relacionadas con la perfusión (ver sección Advertencias y Precauciones Especiales de Uso), así como se debe asegurar la disponibilidad de un equipo de reanimación adecuado. Para consultar las instrucciones de dilución del medicamento antes de la administración, ver sección Instrucciones de Uso, Manipulación y Eliminación.
Modo de Empleo:Instrucciones de uso, manipulación y eliminación: No agitar el vial. Preparar la solución para perfusión usando técnicas asépticas para garantizar la esterilidad de la disolución preparada. Cada vial es para un solo uso. Antes de la dilución se debe comprobar el contenido de los viales para detectar la posible existencia de partículas o cambios de color (la solución debe ser de transparente a ligeramente opalescente e incolora a ligeramente amarilla, sin partículas visibles). Si se identifican partículas o alteraciones del color, el vial se debe descartar. Calcular la dosis y el volumen de Cyramza® necesarios para preparar la solución para perfusión. Los viales contienen 100 mg o 500 mg en solución a 10 mg/ml de Cyramza®. Utilizar únicamente como diluyente cloruro de sodio 9 mg/ml (0.9 %) solución inyectable. En caso de uso de un envase precargado para perfusión intravenosa: Según el volumen de Cyramza® calculado, retirar el volumen correspondiente de solución inyectable de cloruro de sodio 9 mg/ml (0.9 %) del envase precargado de 250 ml para perfusión intravenosa. El paso del volumen de Cyramza® calculado al envase para perfusión intravenosa se debe realizar asépticamente. El volumen total final del envase debe ser 250 ml. El envase se debe invertir cuidadosamente para garantizar una mezcla adecuada. No congelar ni agitar la solución para perfusión. No diluir con otras soluciones o coperfundir con otros medicamentos o electrolitos. En caso de uso de un envase vacío para perfusión intravenosa: El paso de volumen de Cyramza® calculado al envase para perfusión intravenosa vacío se debe realizar asépticamente. Ańadir una cantidad suficiente de solución inyectable de cloruro de sodio 9 mg/ml (0.9 %) al envase para alcanzar un volumen total de 250 ml. El envase se debe invertir cuidadosamente para garantizar una mezcla adecuada. No congelar ni agitar. No diluir con otras soluciones o coperfundir con otros medicamentos o electrolitos. Los medicamentos de administración parenteral se deben examinar visualmente antes de la administración para descartar la presencia de partículas. Si se identifican partículas, el vial se debe descartar. Desechar cualquier porción de Cyramza® remanente en el vial dado que el medicamento no contiene conservantes antimicrobianos. Administrar a través de una bomba de perfusión. Se debe utilizar una vía de perfusión separada con un filtro de entrada de baja unión a proteínas de 0.22 micras para la perfusión y al finalizar la perfusión se debe lavar la vía con solución inyectable de cloruro de sodio 9 mg/ml (0.9 %). La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
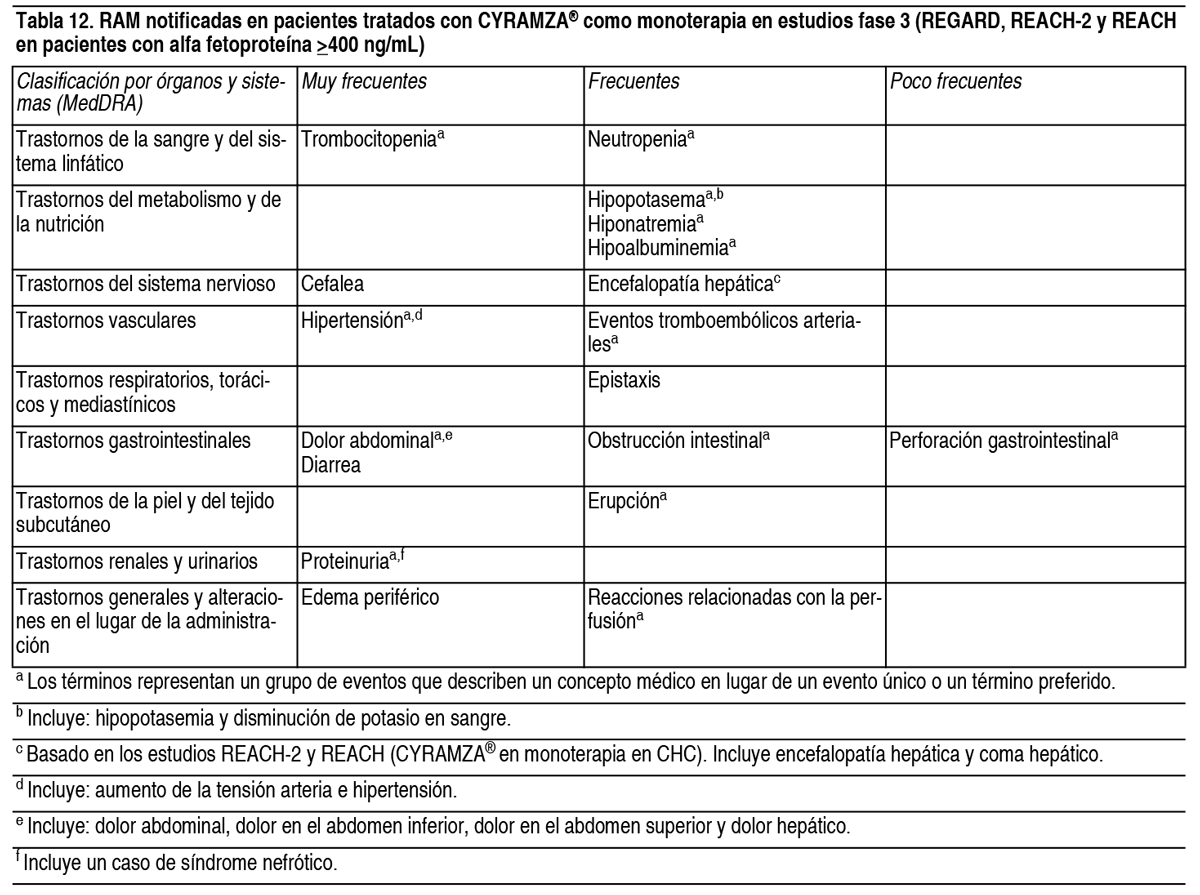
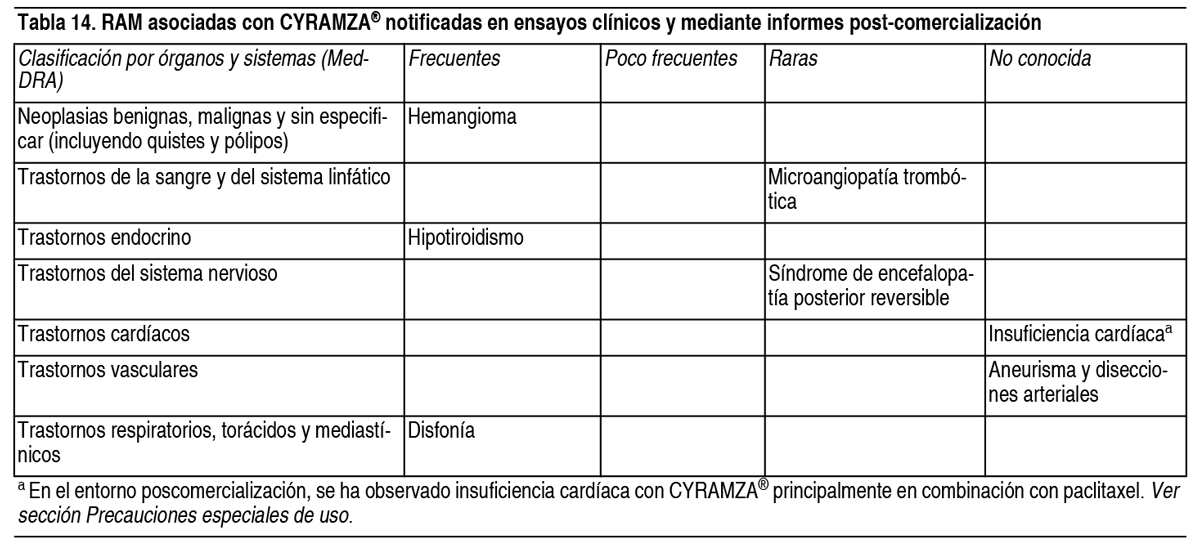
Efectos Colaterales:Resumen del perfil de seguridad: Las reacciones adversas más graves asociadas al tratamiento con Cyramza® (como agente único o en combinación con quimioterapia citotóxica) fueron: Perforación gastrointestinal (ver sección Advertencias y Precauciones Especiales de Uso). Hemorragia gastrointestinal grave (ver sección Advertencias y Precauciones Especiales de Uso). Enfermedad tromboembólica arterial (ver sección Advertencias y Precauciones Especiales de Uso). Síndrome de encefalopatía posterior reversible (ver sección Advertencias y Precauciones Especiales de Uso). Las reacciones adversas más frecuentes observadas en los pacientes tratados con Cyramza® en monoterapia son: Edema periférico, hipertensión, diarrea, dolor abdominal, dolor de cabeza, proteinuria y trombocitopenia. Las reacciones adversas más frecuentes observadas en los pacientes tratados con Cyramza® en combinación con quimioterapia son: Fatiga/astenia, neutropenia, diarrea, epistaxis y estomatitis. Las reacciones adversas más frecuentes observadas en los pacientes tratados con Cyramza® en combinación con erlotinib son: Infecciones, diarrea, hipertensión, estomatitis, proteinuria, alopecia y epistaxis. Tabla de reacciones adversas: Las Tablas 12 y 13 a continuación describen las RAM de los ensayos clínicos fase III controlados con placebo asociados con Cyramza® utilizados tanto en el tratamiento en monoterapia para cáncer gástrico y CHC, como en combinación con diferentes regímenes de quimioterapia o erlotinib para el tratamiento del cáncer gástrico, mCRC y CPNM. Las RAM se incluyen a continuación según la clasificación por órganos y sistemas de MedDRA. La siguiente convención es la utilizada para clasificar la frecuencia en las tablas de RAM: Muy frecuentes (≥ 1/10). Frecuentes (≥ 1/100 a < 1/10). Poco frecuentes (≥ 1/1000 a < 1/100). Raras (≥ 1/10 000 a < 1/1000). Muy raras (< 1/10 000). Frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada grupo de frecuencia, las RAM se presentan en orden decreciente de gravedad.
Las reacciones clínicamente relevantes (incluyendo las de Grado ≥ 3) asociadas a terapia antiangiogénica y observadas en pacientes tratados con ramucirumab durante los ensayos clínicos fueron perforación gastrointestinal, reacciones relacionadas con la perfusión y proteinuria (ver secciones Posología y Modo de Administración y Advertencias y Precauciones Especiales de Uso). Cancer colorrectal: Cyramza®en combinación con FOLFIRI: En el estudio RAISE, en pacientes con mCRC tratados con ramucirumab más FOLFIRI, la RAM más frecuente (≥ 1 %) que llevó a la interrupción de Cyramza® fue proteinuria (1.5 %). Las RAM más frecuentes (≥ 1 %) que llevaron a la interrupción de 1 o más componentes de FOLFIRI fueron neutropenia (12.5 %), trombocitopenia (4.2 %), diarrea (2.3 %) y estomatitis (2.3 %). El componente de FOLFIRI interrumpido de forma más frecuente fue 5-FU en bolo. Reacciones adversas de otras fuentes:
Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Departamento de Farmacovigilancia del laboratorio vía email: fvigilancia@raffo.com.ar o a través de los teléfonos (011) 4509 - 7100 / 7367, y/o del Sistema Nacional de Farmacovigilancia al siguiente link: https://primaryreporting.who-umc.org/AR
Contraindicaciones: Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección Fórmula Cuali-cuantitativa. Para pacientes con CPNM, Cyramza® está contraindicado donde haya cavitación del tumor o afectación tumoral de los grandes vasos sanguíneos (ver sección Advertencias y Precauciones Especiales de Uso).
Advertencias:Trazabilidad: Con objeto de mejorar la trazabilidad de los medicamentos biológicos, el nombre y el número de lote del medicamento administrado deben estar claramente registrados. Enfermedad tromboembólica arterial: En los ensayos clínicos, se han notificado casos graves, ocasionalmente mortales, de enfermedad tromboembólica arterial (arterial thromboembolic events-ATE, por sus siglas en inglés) incluyendo infarto de miocardio, parada cardíaca, accidente cerebrovascular e isquemia cerebral. Cyramza® se debe interrumpir de forma permanente en aquellos pacientes que presenten ATE grave (ver sección Posología y Modo de Administración). Perforaciones Gastrointestinales: Cyramza® es un tratamiento antiangiogénico y puede aumentar el riesgo de perforaciones gastrointestinales. Se han notificado casos de perforación gastrointestinal en pacientes tratados con Cyramza®. El tratamiento con Cyramza® se debe interrumpir de forma permanente en pacientes que presenten perforaciones gastrointestinales (ver sección Posología y Modo de Administración). Hemorragia grave: Cyramza® es un tratamiento antiangiogénico y puede aumentar el riesgo de hemorragia grave. El tratamiento con Cyramza® se debe interrumpir de forma permanente en aquellos pacientes que presenten hemorragia de Grado 3 o 4 (ver sección Posología y Modo de Administración). En aquellos pacientes con enfermedades que predispongan al sangrado y en aquellos tratados con anticoagulantes u otros medicamentos que incrementan el riesgo hemorrágico se deben monitorizar los parámetros de hemograma y coagulación. Para pacientes con CHC con evidencia de hipertensión portal o antecedentes de sangrado de varices esofágicas, se debe realizar un cribado y tratar las varices esofágicas con el tratamiento adecuado antes de empezar el tratamiento con Cyramza®. En pacientes con cáncer gástrico tratados con Cyramza® en combinación con paclitaxel y en pacientes con mCRC tratados con Cyramza® en combinación con FOLFIRI, se han notificado hemorragias gastrointestinales graves incluyendo casos mortales. Hemorragia pulmonar en CPNM: Los pacientes con histología escamosa tienen un riesgo mayor de desarrollar hemorragia pulmonar grave, sin embargo, en el estudio REVEL en pacientes tratados con Cyramza® con histología escamosa, no se observó un exceso de hemorragia pulmonar Grado 5. En los ensayos clínicos se excluyeron aquellos pacientes con CPNM con hemorragia pulmonar reciente (> 2.5 ml o sangre roja brillante) así como pacientes con evidencia basal de cavitación tumoral, independientemente de la histología, o aquellos con evidencia de invasión tumoral o confinamiento de grandes vasos sanguíneos (ver sección Contraindicaciones). Los pacientes que recibieron cualquier tipo de tratamiento anticoagulante fueron excluidos del estudio REVEL en CPNM y los pacientes que estaban recibiendo tratamiento crónico con fármacos antiinflamatorios no esteroideos o agentes antiplaquetarios fueron excluidos de los ensayos clínicos REVEL y RELAY en CPNM. Se permitió el uso de Aspirina a dosis de hasta 325 mg/día (ver sección Propiedades Farmacodinámicas). Reacciones relacionadas con la perfusión: Se han notificado reacciones relacionadas con la perfusión en ensayos clínicos con Cyramza®. La mayoría de los casos ocurrieron durante o tras una primera o segunda perfusión de Cyramza®. Durante la perfusión, los pacientes deben ser monitorizados ante la posible aparición de signos y síntomas de hipersensibilidad. Estos incluyeron temblores, espasmos/dolor de espalda, dolor y/u opresión torácica, escalofríos, rubor, disnea, sibilancias, hipoxia, y parestesias. En casos graves, estos incluyeron broncoespasmo, taquicardia supraventricular e hipotensión. El tratamiento con Cyramza® se debe interrumpir de forma inmediata y permanente en pacientes que presenten IRR de Grado 3 o 4 (ver sección Posología y Modo de Administración). Hipertensión: Se notificó un aumento en la incidencia de casos de hipertensión grave en pacientes que recibieron Cyramza® comparado con placebo. En la mayoría de los casos, la hipertensión se manejó utilizando un tratamiento antihipertensivo estándar. Los pacientes con hipertensión no controlada fueron excluidos de los ensayos: no se debe comenzar el tratamiento con Cyramza® en estos pacientes hasta que la hipertensión preexistente se controle. La tensión arterial se debe monitorizar en los pacientes tratados con Cyramza®. En caso de hipertensión grave, el tratamiento con Cyramza® se debe interrumpir temporalmente hasta que se controle con medicación. En caso de hipertensión médicamente significativa que no pueda ser controlada con antihipertensivos, el tratamiento con Cyramza® se debe interrumpir de forma permanente (ver sección Posología y Modo de Administración). Síndrome de encefalopatía posterior reversible: Raramente se han notificado casos de síndrome de encefalopatía posterior reversible (PRES, por sus siglas en inglés), incluidos casos mortales, en pacientes que reciben Cyramza®. Los síntomas de PRES pueden incluir convulsiones, dolor de cabeza, náuseas/vómitos, ceguera o alteración de la conciencia, con o sin hipertensión asociada. El diagnóstico de PRES se puede confirmar mediante imágenes cerebrales (por ejemplo, imágenes de resonancia magnética). Interrumpa el tratamiento de Cyramza® en pacientes que experimentan PRES. No se conoce la seguridad de reiniciar Cyramza® en pacientes que desarrollan PRES y se recuperan. Aneurisma y disecciones arteriales: El uso de inhibidores de la vía VEGF en pacientes con o sin hipertensión puede promover la formación de aneurismas y/o de disecciones arteriales. Antes de iniciar la administración de Cyramza®, este riesgo se debe evaluar de forma cuidadosa en pacientes con factores de riesgo como la hipertensión o antecedentes de aneurisma. Dificultad en la curación de heridas: El impacto de Cyramza® no se ha evaluado en pacientes con heridas no curadas o graves. En un ensayo llevado a cabo en animales, Cyramza® no dificultó la curación de heridas. Sin embargo, dado que Cyramza® es un tratamiento antiangiogénico y puede potencialmente afectar de forma negativa a la curación de heridas, el tratamiento con Cyramza® se debe interrumpir durante al menos las 4 semanas previas a una cirugía programada. La decisión de reanudar el tratamiento con Cyramza® tras la intervención quirúrgica debe basarse en el juicio clínico para lograr una adecuada curación de la herida. Si durante el tratamiento un paciente desarrolla complicaciones en la curación de heridas, Cyramza® se debe interrumpir hasta que la herida esté completamente curada (ver sección Posología y Modo de Administración). Insuficiencia hepática: Cyramza® se debe utilizar con precaución en pacientes con cirrosis hepática grave (Child-Pugh B o C), cirrosis con encefalopatía hepática, ascitis debida a la cirrosis clínicamente significativa o síndrome hepatorrenal. Existen muy pocos datos de eficacia y seguridad disponibles en estos pacientes. Cyramza® se debe utilizar únicamente en estos pacientes si se considera que el beneficio potencial del tratamiento supera el riesgo potencial de progresión a fallo hepático. En pacientes con CHC, se notificó encefalopatía hepática en una mayor proporción en los pacientes tratados con Cyramza® en comparación con los pacientes tratados con placebo (ver sección Reacciones Adversas). Se debe vigilar a los pacientes para detectar signos clínicos y síntomas de encefalopatía hepática. Cyramza® se debe interrumpir de forma permanente en caso de encefalopatía hepática o síndrome hepatorrenal (ver sección Posología y Modo de Administración). Insuficiencia cardíaca: En los datos agrupados de los ensayos clínicos con ramucirumab, se notificó insuficiencia cardíaca con una incidencia numéricamente más alta en pacientes que recibieron ramucirumab en combinación con diversos regímenes de quimioterapia, o erlotinib, en comparación con quimioterapia o erlotinib solo. Este aumento de la incidencia no se observó en los pacientes que recibieron ramucirumab en comparación con placebo en los ensayos clínicos como agente único. En el entorno poscomercialización, se ha observado insuficiencia cardíaca con ramucirumab, principalmente en combinación con paclitaxel. Se debe monitorizar a los pacientes para detectar signos y síntomas clínicos de insuficiencia cardíaca durante el tratamiento, y se debe considerar la suspensión del tratamiento si se desarrollan signos y síntomas clínicos de insuficiencia cardíaca. Ver sección Reacciones Adversas. Fístulas: Los pacientes tratados con Cyramza® pueden presentar un riesgo mayor de desarrollar fístulas. Se debe interrumpir el tratamiento con Cyramza® en pacientes que desarrollan una fístula (ver sección Posología y Modo de Administración). Proteinuria: En pacientes que recibieron Cyramza® se notificó un aumento en la incidencia de proteinuria comparado con los que recibieron placebo. Durante el tratamiento con Cyramza® se debe controlar el desarrollo o empeoramiento de la proteinuria en el paciente. Si los niveles de proteína en orina son ≥ 2+ en una tira reactiva de orina, se debe recoger una muestra de orina de 24 horas. El tratamiento con Cyramza® se debe interrumpir temporalmente en caso de niveles de proteína en orina ≥ 2 g/24 horas. Una vez que los niveles de proteínas en orina se hayan restablecido a valores < 2 g/24 horas, se debe reiniciar el tratamiento con una dosis reducida. Si reaparecen niveles de proteína en orina ≥ 2 g/24 horas, se recomienda una segunda reducción de la dosis. Si el nivel de proteinuria es > 3 g/24 horas o existe síndrome nefrótico, el tratamiento con Cyramza® se debe interrumpir de forma permanente (ver sección Posología y Modo de Administración). Estomatitis: Se notificó un aumento de la incidencia de estomatitis en pacientes que recibieron Cyramza® en combinación con quimioterapia comparado con pacientes tratados con placebo más quimioterapia. Si la estomatitis aparece, el tratamiento sintomático se debe instaurar lo antes posible. Insuficiencia renal: Los datos de seguridad disponibles para pacientes con insuficiencia renal grave (aclaramiento de creatinina 15 a 29 ml/min) tratados con Cyramza® son limitados (ver las secciones Posología y Modo de Administración y Propiedades Farmacocinéticas). Pacientes de edad avanzada con CPNM: Se ha observado una tendencia a menor eficacia conforme aumenta la edad del paciente que recibe Cyramza® más docetaxel para el tratamiento de CPNM avanzado con progresión de la enfermedad tras quimioterapia basada en platino (ver sección Propiedades Farmacodinámicas). Antes del comienzo de tratamiento en pacientes de edad avanzada, se debe evaluar cuidadosamente las comorbilidades, el PS y la potencial tolerabilidad a la quimioterapia (ver las secciones Posología y Modo de Administración y Propiedades Farmacodinámicas). Con el uso de Cyramza® en combinación con erlotinib para el tratamiento en primera línea de CPNM con mutaciones activadoras de EGFR, los pacientes de 70 o más ańos en comparación con los pacientes menores de 70 ańos, experimentaron una mayor incidencia de efectos adversos de grado ≥ 3 y de todos los efectos adversos graves de cualquier grado. Sodio: Este medicamento contiene menos de 1 mmol de sodio (23 mg) en cada vial de 10 ml; esto es, esencialmente «exento de sodio». Este medicamento contiene aproximadamente 85 mg de sodio en cada vial de 50 ml. Esto equivale al 4 % de la ingesta máxima diaria de 2 g de sodio recomendada por la OMS para un adulto.
Precauciones:Fertilidad, embarazo y lactancia: Mujeres en edad fértil/anticoncepción en mujeres: Se debe aconsejar a las mujeres en edad fértil que eviten el embarazo durante el tratamiento con Cyramza® y deben ser informadas del dańo potencial al embarazo y al feto. Las mujeres en edad fértil deben usar métodos anticonceptivos eficaces durante y hasta los 3 meses posteriores a la última dosis del tratamiento con Cyramza®. Embarazo: No existen datos del uso de Cyramza® en mujeres embarazadas. Los estudios en animales son insuficientes con respecto a la toxicidad reproductiva (ver sección Datos Preclínicos sobre Seguridad). La angiogénesis es crítica durante el mantenimiento del embarazo y el desarrollo fetal, por lo que la inhibición de la angiogénesis tras la administración de Cyramza® puede provocar reacciones adversas sobre el embarazo, incluyendo al feto. Solo se debe utilizar Cyramza® si el beneficio potencial a la madre justifica el riesgo potencial durante el embarazo. Si la paciente llega a quedarse embarazada durante el tratamiento con Cyramza®, se le debe informar del riesgo potencial de mantener el embarazo y del riesgo para el feto. No se recomienda Cyramza® durante el embarazo ni en mujeres en edad fértil que no usan anticonceptivos. Lactancia: Se desconoce si Cyramza® se excreta en la leche materna. Se espera que la excreción en la leche y la absorción oral sean bajas. Dado que no se puede excluir el riesgo en lactantes, la lactancia se debe interrumpir durante el tratamiento con Cyramza® y durante al menos 3 meses tras la última dosis. Fertilidad: No existen datos del efecto de Cyramza® sobre la fertilidad humana. Según estudios en animales, se considera probable que la fertilidad en mujeres se pueda ver comprometida durante el tratamiento con Cyramza® (ver sección Datos Preclínicos sobre Seguridad). Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de Cyramza® sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Si el paciente sufre síntomas que afectan su capacidad de reacción o su habilidad para concentrarse, se recomienda que no conduzca o utilice máquinas hasta que los efectos disminuyan.
Interacciones Medicamentosas: No se han observado interacciones farmacológicas entre Cyramza® y paclitaxel. La farmacocinética de paclitaxel no se vio afectada cuando se administró de forma conjunta con Cyramza® y la farmacocinética de Cyramza® no se vio afectada cuando se administró de forma conjunta con paclitaxel. La farmacocinética de irinotecán y la de su metabolito activo, SN-38, no se vieron afectadas cuando se administró junto con Cyramza®. La farmacocinética de docetaxel o erlotinib no se vio afectada cuando se administró junto con Cyramza®.
Sobredosificación: No existen datos de sobredosis en humanos. En un estudio fase 1, se administró Cyramza® hasta 10 mg/kg cada 2 semanas sin alcanzar la dosis máxima tolerada. En caso de sobredosis, se debe utilizar un tratamiento sintomático. En Argentina: Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría «Dr. Ricardo Gutiérrez»: (011) 4962 6666 / 2247. Hospital «Dr. Alejandro Posadas»: (011) 4658 7777 / 4654 6648. Optativamente otros Centros de Intoxicaciones.
Incompatibilidades: Cyramza® no debe administrarse o mezclarse con soluciones de dextrosa. Este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección Instrucciones de Uso, Manipulación y Eliminación.
Conservación:Precauciones especiales para el almacenamiento: Conservar los viales en refrigeración a una temperatura de 2 °C a 8 °C hasta el momento de uso. Conservar en el envase original protegido de la luz. No congelar ni agitar el vial. Viales en uso (solución diluida y preparada): La solución diluida y preparada se debe utilizar inmediatamente, ya que no contiene conservantes. Si no es utilizada inmediatamente, conservar en refrigeración entre 2 °C y 8 °C hasta 24 horas o a temperatura ambiente no mayor a 30 °C por 4 horas (si la dilución se hizo utilizando técnicas asépticas aceptables). Si la solución diluida está refrigerada, permita que alcance la temperatura ambiente antes de la administración. El tiempo de conservación incluye la duración de la perfusión. No congelar ni agitar la solución diluida y preparada.
Observaciones: Manténgase fuera del alcance de los nińos. No usar este medicamento después de la fecha de vencimiento indicada en el envase.
Presentaciones: Cyramza® se presenta en forma de solución estéril sin conservantes en viales unidosis. 100 mg/10 ml (10 mg/ml), en estuche individual. 500 mg/50 ml (10 mg/ml), en estuche individual.
 Laboratorio
Laboratorio