Aparato Circulatorio: Vasodilatadores Centrales y Periféricos
Sangre: Anticoagulantes Antitrombóticos
Información farmacológica
Composición:Corionil®50 mg: Cada comprimido contiene: Cilostazol 50 mg. Excipientes: Lactosa, Povidona, Glicolato Sódico de Almidón, Ludipress y Estearato de Magnesio c.s. Corionil®100 mg: Cada comprimido contiene: Cilostazol 100 mg. Excipientes: Lactosa, Povidona, Glicolato Sódico de Almidón, Ludipress y Estearato de Magnesio c.s.
Acción Terapéutica: El cilostazol es un derivado de la quinolinona que inhibe a la fosfodiesterasa III (PDE III). La elevación resultante del AMP cíclico (AMPc) a nivel intracelular inhibe la agregación plaquetaria y produce vasodilatación.
Indicaciones: Corionil® está indicado para la reducción de los síntomas de la claudicación intermitente.
Propiedades:Acción farmacológica: El mecanismo de acción del cilostazol sobre los síntomas de la claudicación intermitente no es totalmente conocido. El cilostazol y varios de sus metabolitos son inhibidores de la PDE III específica para el AMPc. La inhibición de la actividad de la PDE suprime la degradación del AMPc, dando como resultado un aumento de AMPc en plaquetas y vasos sanguíneos, lo que conduce a una inhibición de la agregación plaquetaria y a la vasodilatación, respectivamente. El cilostazol inhibe de manera reversible la agregación plaquetaria inducida por varios estímulos, los que incluyen a la trombina, el ADP, el colágeno, el ácido araquidónico, la adrenalina y el estrés. El cilostazol administrado en dosis de 100 mg dos veces al día durante 12 semanas produjo una reducción de los triglicéridos plasmáticos de alrededor del 15% y un aumento del colesterol HDL de aproximadamente un 10%. Efectos cardiovasculares: El cilostazol produce modificaciones tanto a nivel de los lechos vasculares como de la función cardiovascular. Produce una vasodilatación no homogénea, con mayor dilatación de los lechos femorales que de los vertebrales, carotídeos o mesentéricos superiores. Las arterias renales no son sensibles a los efectos del cilostazol. En perros y monos, el cilostazol aumentó la frecuencia cardíaca, la contractilidad miocárdica y el flujo coronario, así como la automaticidad ventricular, como es de esperar con un inhibidor de la PDE III. La contractilidad del ventrículo izquierdo y la conducción AV aumentaron con las dosis requeridas para inhibir la agregación plaquetaria. En humanos, la frecuencia cardíaca aumentó en forma proporcional a la dosis, con un promedio de 5.1 y 7.4 latidos por minuto en pacientes tratados con 50 y 100 mg 2 veces al día, respectivamente. En pacientes evaluados con monitoreo Holter, un mayor número de pacientes tratados con cilostazol experimentaron aumento de extrasístoles ventriculares y episodios de taquicardia ventricular no sostenida que los pacientes tratados con placebo, pero este aumento no estuvo relacionado con la dosis. Farmacocinética: El cilostazol se absorbe luego de la administración oral. La absorción es incrementada mediante una comida rica en grasas, con una elevación de la concentración plasmática máxima (Cmáx) de aproximadamente un 90% y un aumento del 25% en el área bajo la curva de concentración plasmática-tiempo (AUC). No se conoce la biodisponibilidad absoluta. El cilostazol es ampliamente metabolizado por las enzimas hepáticas del citocromo P-450, principalmente 3A4, y los metabolitos se excretan en su gran mayoría en la orina. Dos metabolitos son activos, y uno de ellos parece aportar un 50% de la actividad farmacológica (inhibición de la PDE III) luego de la administración de cilostazol. La farmacocinética es aproximadamente proporcional a la dosis. El cilostazol y sus metabolitos activos tienen una vida media de eliminación aparente de 11 a 13 horas. El cilostazol y sus metabolitos activos se acumulan alrededor de 2 veces con la administración crónica y alcanzan las concentraciones en sangre del estado estacionario en unos pocos días. La farmacocinética del cilostazol y de sus 2 principales metabolitos activos es similar en sujetos sanos normales y en pacientes con claudicación intermitente debida a arteriopatía periférica (AP). El cilostazol se une en un 95 a 98% a las proteínas plasmáticas, predominantemente a la albúmina. La unión porcentual promedio del 3,4-dehidrocilostazol es de 97.4% y la del 4´-trans-hidroxicilostazol de 66%. En la insuficiencia hepática leve no está afectada la unión a proteínas. La fracción libre de cilostazol fue un 27% mayor en sujetos con insuficiencia renal que en voluntarios normales. El desplazamiento de las proteínas plasmáticas del cilostazol por eritromicina, quinidina, warfarina y omeprazol no fue clínicamente significativo. El cilostazol se elimina predominantemente por metabolización y subsiguiente excreción urinaria de los metabolitos. Sobre la base de estudios in vitro, las principales isoenzimas involucradas en el metabolismo del cilostazol son CYP3A4 y, en menor grado, CYP2C19. Se desconoce la enzima responsable del metabolismo del 3,4-dehidrocilostazol, el más activo de los metabolitos. Luego de la administración oral de 100 mg de cilostazol radiomarcado, un 56% de la actividad plasmática total correspondió a cilostazol, 15% a 3,4-dehidrocilostazol (4 a 7 veces más activo que el cilostazol) y 4% a 4´-transhidroxicilostazol (1/5 de la actividad del cilostazol). La vía principal de eliminación fue la orina (74%) y el resto se excretó en las heces (20%). No se excretó una cantidad medible de cilostazol sin cambios en la orina y menos del 2% de la dosis fue excretada como 3,4-dehidrocilostazol. Alrededor del 30% de la dosis fue excretada en la orina como 4´-transhidroxicilostazol y el resto como metabolitos, ninguno de los cuales excedió el 5%. No hubo evidencia de inducción de enzimas microsomales hepáticas. Poblaciones especiales: Edad y sexo: la depuración oral del cilostazol y sus metabolitos, ajustada por el peso corporal, no cambió significativamente con el sexo y/o la edad dentro del rango de 50 a 80 ańos. Fumadores: los análisis de farmacocinética poblacional sugieren que el tabaquismo disminuye la exposición al cilostazol en alrededor del 20%. Insuficiencia hepática: la farmacocinética del cilostazol y de sus metabolitos fue similar en pacientes con insuficiencia hepática leve en comparación con sujetos normales. No se han estudiado pacientes con insuficiencia hepática moderada o severa. Insuficiencia renal: la actividad farmacológica total del cilostazol y sus metabolitos fue similar en sujetos con insuficiencia renal leve a moderada y en sujetos normales. La insuficiencia renal severa aumenta los niveles de los metabolitos y altera la unión a proteínas de la droga madre y de los metabolitos. Sin embargo, la actividad farmacológica esperada, sobre la base de las concentraciones plasmáticas, y la potencia inhibitoria relativa de la PDE III de la droga madre y de los metabolitos se modificaron muy poco. No se han estudiado pacientes sometidos a diálisis, pero es improbable que el cilostazol pueda ser removido en forma eficiente por este procedimiento, debido a su elevada ligadura proteica (95 a 98%). Interacciones farmacocinéticas y farmacodinámicas: (véase Posología): El cilostazol puede tener interacciones farmacodinámicas con otros inhibidores de la función plaquetaria e interacciones farmacocinéticas debido a los efectos de otras drogas sobre su metabolización por el CYP3A4 o el CYP2C19. El cilostazol no parece inhibir al CYP3A4. Aspirina: la administración a corto plazo (≤ 4 días) de aspirina junto con cilostazol produjo un aumento del 23 al 35% en la inhibición de la agregación plaquetaria inducida por ADP ex vivo en comparación con la aspirina sola; no hubo impacto clínicamente significativo sobre el TP, KPTT o el tiempo de sangría en comparación con aspirina sola, ni un efecto sinérgico o aditivo sobre la agregación plaquetaria inducida por ácido araquidónico. Se desconocen los efectos de la administración conjunta a largo plazo en la población general. En ensayos clínicos a doble ciego, randomizados, controlados con placebo, en los que se empleó aspirina (75 - 81 mg diarios durante 137 días) conjuntamente con cilostazol (325 mg diarios durante 54 días), no hubo un aumento evidente en la incidencia de efectos adversos hemorrágicos en pacientes que tomaban aspirina y cilostazol en comparación con los pacientes que tomaban placebo y dosis equivalentes de aspirina. Warfarina: las isoenzimas del citocromo P-450 involucradas en el metabolismo de la R-warfarina son 3A4, 1A2 y 2C19, en tanto que en el metabolismo de la S-warfarina, interviene el CYP2C9. El cilostazol no inhibe el metabolismo ni los efectos farmacológicos (TP, KPTT, tiempo de sangría o agregación plaquetaria) de la R- y S-warfarina luego de una única dosis de 25 mg de warfarina. Se desconoce el efecto de dosis múltiples de warfarina y cilostazol sobre la farmacocinética y farmacodinamia de ambas drogas. Omeprazol: la administración conjunta con omeprazol no afectó significativamente la metabolización del cilostazol, pero la exposición sistémica al 3,4-dehidrocilostazol aumentó en un 69%, probablemente como resultado de la inhibición potente del CYP2C19 por el omeprazol. Eritromicina y otros antibióticos macrólidos: la eritromicina es un inhibidor relativamente potente del CYP3A4. La administración conjunta de 500 mg de eritromicina cada 8 horas con una única dosis de 100 mg de cilostazol aumentó la Cmáx del cilostazol en un 47% y el AUC en un 73%. La inhibición del metabolismo del cilostazol por la eritromicina aumentó el AUC del 4´-trans- hidroxicilostazol en un 141%. Es de esperar que otros antibióticos macrólidos tengan un efecto similar. Diltiazem: se ha demostrado que el diltiazem, un inhibidor moderado del CYP3A4, aumenta las concentraciones plasmáticas del cilostazol aproximadamente un 53%, según un análisis farmacocinético poblacional. Quinidina: la administración concomitante de quinidina con una única dosis de 100 mg de cilostazol no alteró la farmacocinética de este último. Inhibidores potentes del CYP3A4: los inhibidores potentes del CYP3A4, como el ketoconazol, itraconazol, fluconazol, miconazol, fluvoxamina, fluoxetina, nefazodona y sertralina no han sido estudiados en combinación con cilostazol, pero es de esperar que causen un mayor aumento de los niveles plasmáticos del cilostazol y sus metabolitos que la eritromicina. Lovastatin: la administración concomitante de una dosis única de 80 mg de lovastatin con el cilostazol no produjo aumentos clínicamente significativos en las concentraciones plasmáticas de lovastatin y su metabolito hidroxiácido en estado estacionario.
Posología: La dosis recomendada de Corionil® es de 100 mg 2 veces al día, tomados como mínimo 1/2 hora antes o 2 horas después del desayuno y de la cena. Los pacientes pueden responder a las 2 a 4 semanas de iniciar la terapia, pero puede ser necesario el tratamiento durante 12 semanas antes de que se experimente mejoría. Se debe considerar una dosis de 50 mg 2 veces al día durante la administración concomitante de inhibidores del CYP3A4 como ketoconazol, itraconazol, eritromicina, diltiazem y de inhibidores del CYP2C19 como el omeprazol. El CYP3A4 también es inhibido por el jugo de pomelo. Debido a que la magnitud y el curso temporal de esta interacción no ha sido investigada aún, los pacientes que toman Corionil® deben evitar consumir jugo de pomelo. Interrupción del tratamiento: los datos disponibles sugieren que se puede reducir la dosis de cilostazol o interrumpirlo sin que haya efecto rebote (hiperagregabilidad plaquetaria).
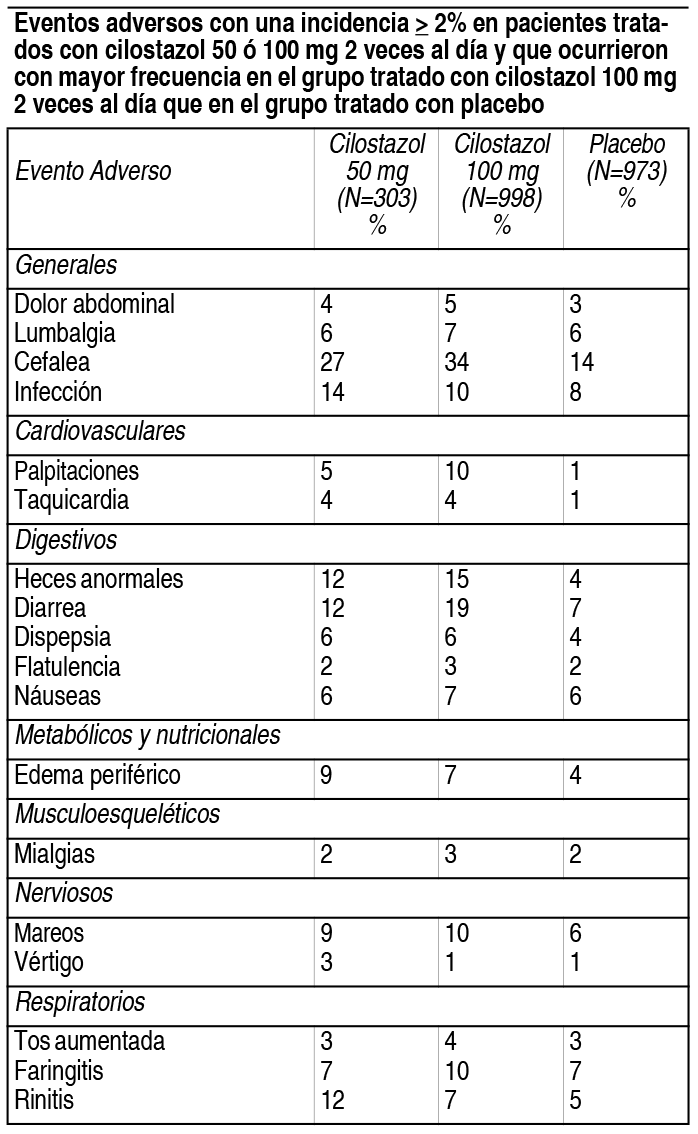
Efectos Colaterales: Los eventos adversos asociados al uso del cilostazol se evaluaron en estudios clínicos controlados con placebo, que incluyeron a 2.274 pacientes, de los cuales 1.301 recibieron dosis de 50 ó 100 mg de cilostazol 2 veces al día y 973 recibieron placebo, con una duración promedio del tratamiento de 127 días para los pacientes que recibieron cilostazol y 134 días para los pacientes que recibieron placebo. El único evento adverso que resultó en la interrupción de la terapia en ≥ 3% de los pacientes tratados con cilostazol 50 ó 100 mg 2 veces al día fue la cefalea, que ocurrió con una incidencia de 1.3%, 3.5% y 0.3% en pacientes tratados con cilostazol 50 mg, cilostazol 100 mg y placebo, respectivamente. Otras causas frecuentes de interrupción del tratamiento fueron palpitaciones y diarrea, ambas con una frecuencia de 1.1% con cilostazol (ambas dosis) contra 0.1% con placebo. Los eventos adversos informados con más frecuencia, que ocurrieron en ≥ 2% de los pacientes tratados con cilostazol 50 ó 100 mg 2 veces al día, se muestran en la tabla. Otros eventos adversos observados con una incidencia ≥ 2%, pero que ocurrieron en el grupo placebo como mínimo con la misma frecuencia que en el grupo de 100 mg de cilostazol 2 veces al día, fueron: astenia, hipertensión, vómitos, calambres en las piernas, hiperestesia, parestesias, disnea, rash, hematuria, infección urinaria, síndrome gripal, angina de pecho, artritis y bronquitis. Eventos adversos con una incidencia ≥ 2% en pacientes tratados con cilostazol 50 ó 100 mg 2 veces al día y que ocurrieron con mayor frecuencia en el grupo tratado con cilostazol 100 mg 2 veces al día que en el grupo tratado con placebo:
Los eventos adversos menos frecuentes (< 2%) experimentados por pacientes bajo tratamiento con cilostazol 50 ó 100 mg 2 veces al día y que ocurrieron con mayor frecuencia en el grupo tratado con 100 mg de cilostazol que en el grupo placebo, independientemente de que la relación con la droga haya sido o no establecida, se enumeran a continuación:Generales: escalofríos, edema facial, fiebre, edema generalizado, malestar general, rigidez de cuello, dolor pélvico, hemorragia retroperitoneal. Cardiovasculares: fibrilación auricular, aleteo auricular, infarto cerebral, isquemia cerebral, insuficiencia cardíaca congestiva, paro cardíaco, hemorragia, hipotensión, infarto de miocardio, arritmia nodal, hipotensión postural, taquicardia supraventricular, síncope, venas varicosas, vasodilatación, extrasístoles ventriculares, taquicardia ventricular. Digestivos: anorexia, colelitiasis, colitis, úlcera duodenal, duodenitis, hemorragia esofágica, esofagitis, aumento de gamaglutamil transpeptidasa, gastritis, gastroenteritis, hemorragia gingival, hematemesis, melena, úlcera péptica, abceso periodóntico, hemorragia rectal, úlcera de estómago, edema de lengua. Endocrinos: diabetes mellitus. Hemáticos y linfáticos: anemia, equimosis, anemia ferropénica, policitemia, púrpura. Metabólicos y nutricionales: aumento de creatinina, gota, hiperlipemia, hiperuricemia. Musculoesqueléticos: artralgias, dolor óseo, bursitis. Nerviosos: ansiedad, insomnio, neuralgia. Respiratorios: asma, epistaxis, hemoptisis, neumonía, sinusitis. Piel y apéndices: piel seca, forunculosis, hipertrofia cutánea, urticaria. Sentidos especiales: ambliopía, conjuntivitis, diplopía, otalgia, hemorragia ocular, hemorragia retiniana, acúfenos. Urogenitales: albuminuria, cistitis, micción frecuente, hemorragia vaginal, vaginitis.
Contraindicaciones: Corionil® está contraindicado en pacientes con hipersensibilidad conocida o sospechada a cualquiera de sus componentes. Dado que el cilostazol y varios de sus metabolitos son inhibidores de la fosfodiesterasa III y que varias drogas con este efecto farmacológico han causado una disminución de la supervivencia en comparación con placebo en pacientes con insuficiencia cardíaca congestiva clase III-IV, Corionil® está contraindicado en pacientes con insuficiencia cardíaca congestiva de cualquier severidad.
Precauciones: Corionil® está contraindicado en pacientes con insuficiencia cardíaca congestiva. Se desconocen los efectos a largo plazo de los inhibidores de la PDE III (incluyendo al cilostazol) en pacientes sin insuficiencia cardíaca congestiva. En ensayos controlados con placebo de 3 a 6 meses de duración realizados con pacientes relativamente estables (sin infarto de miocardio o accidente cerebrovascular reciente, sin dolor en reposo u otros signos de enfermedad rápidamente progresiva), la mortalidad fue de 0.7% en el grupo placebo y 0.8% en el grupo de cilostazol. No hay datos con respecto al riesgo a corto o largo plazo en pacientes con cardiopatía subyacente más severa. Uso con Clopidogrel: actualmente no hay información disponible con respecto a la seguridad y la eficacia del uso concomitante de cilostazol y clopidogrel, una droga inhibidora de la agregación plaquetaria indicada para pacientes con arteriopatía periférica. Información para los pacientes: Se debe advertir a los pacientes: Que tomen Corionil® por lo menos 1/2 hora antes o 2 horas después de las comidas. Que los efectos de Corionil® sobre los síntomas de la claudicación intermitente pueden no ser inmediatos. Si bien el paciente puede experimentar una mejoría en 2 a 4 semanas luego de iniciar la terapia, puede ser necesario el tratamiento aún durante 12 semanas para obtener un beneficio. Que no hay certeza respecto al riesgo cardiovascular con el uso a largo plazo o en pacientes con cardiopatía severa subyacente. Insuficiencia hepática: no se ha estudiado el uso de cilostazol en pacientes con insuficiencia hepática moderada a severa. Carcinogénesis, mutagénesis, deterioro de la fertilidad: la administración de cilostazol a través de la dieta a ratas y ratones de ambos sexos durante hasta 104 semanas, en dosis de hasta 500 mg/kg/día en ratas y 1.000 mg/kg/día en ratones, no revelaron evidencia de potencial carcinogénico. Las dosis máximas administradas tanto a las ratas como a los ratones fueron, sobre la base de la exposición sistémica, menores a la exposición humana con la dosis máxima recomendada. El cilostazol dio resultado negativo en los ensayos de mutación genética bacteriana, de reparación de DNA bacteriano, de mutación genética en células de mamíferos y en el ensayo in vivo de aberraciones cromosómicas en médula ósea murina. Se asoció, sin embargo, con un aumento significativo de aberraciones cromosómicas en el ensayo in vitro con células de ovario de hámster chino. El cilostazol no afectó la fertilidad ni el apareamiento de ratas macho y hembra en dosis tan altas como 1.000 mg/kg/día. A esta dosis, la exposición sistémica (AUC) al cilostazol libre fue menos de 1.5 veces y alrededor de 5 veces (machos y hembras, respectivamente) la exposición sistémica humana con la dosis máxima recomendada. Embarazo: Categoría C. En un estudio de toxicidad sobre el desarrollo en ratas, la administración oral de 1.000 mg/kg/día de cilostazol se asoció con disminución del peso fetal y un aumento en la incidencia de anomalías cardiovasculares, renales y esqueléticas (anomalías del septum interventricular, del cayado aórtico y de la arteria subclavia; dilatación de la pelvis renal; 14Ş costilla; y retardo de la osificación). Con esta dosis, la exposición sistémica al cilostazol libre en ratas no preńadas fue alrededor de 5 veces la exposición humana a la dosis máxima recomendada. También se observó un aumento en la incidencia de defectos del septum interventricular y osificación retardada con 150 mg/kg/día (5 veces la exposición sistémica humana). En un estudio de toxicidad sobre el desarrollo en conejos, se observó un aumento en la incidencia de retardo de la osificación del esternón con dosis de 150 mg/kg/día. En conejas no preńadas, la exposición sistémica al cilostazol libre con la dosis de 150 mg/kg/día fue considerablemente menor a la que se observa en humanos con la dosis máxima recomendada y la exposición al 3,4-dehidrocilostazol fue apenas detectable. Cuando se administró cilostazol a ratas durante la última etapa de la preńez y durante la lactancia, se observó un aumento en la incidencia de muerte fetal y disminución del peso de las crías con dosis de 150 mg/kg/día (5 veces la exposición humana con la dosis máxima recomendada). No hay estudios adecuados y bien controlados en mujeres embarazadas. Lactancia: se ha informado de la transferencia de cilostazol a la leche en animales de experimentación (ratas). Debido al riesgo potencial para el lactante, se debe tomar la decisión, en el caso que corresponda, de interrumpir la lactancia o el tratamiento con Corionil®. Uso pediátrico: no se han establecido la seguridad y la eficacia de Corionil® en pacientes pediátricos. Uso geriátrico: en los estudios clínicos con cilostazol (n = 2.274), el 56% de los pacientes tenían 65 ańos o más, mientras que el 16% tenían 75 ańos o más. No se observaron diferencias globales en la seguridad o eficacia entre estos individuos y los más jóvenes, y la experiencia clínica informada no identificó diferencias en la respuesta entre los individuos ancianos y los más jóvenes, pero no se puede excluir una mayor sensibilidad de algunos pacientes ancianos. Los estudios farmacocinéticos no han descubierto ningún efecto relacionado con la edad sobre la absorción, la distribución, el metabolismo y la eliminación del cilostazol y sus metabolitos.
Interacciones Medicamentosas: Dado que el cilostazol se metaboliza extensamente por las isoenzimas del citocromo P-450, se debe tener precaución cuando se administra Corionil® conjuntamente con inhibidores del CYP3A4 como el ketoconazol y la eritromicina o inhibidores del CYP2C19 como el omeprazol. Los estudios farmacocinéticos han demostrado que el omeprazol y la eritromicina aumentan significativamente la exposición sistémica al cilostazol y/o a sus metabolitos principales. Los estudios farmacocinéticos poblacionales mostraron mayores concentraciones de cilostazol en los pacientes tratados conjuntamente con diltiazem, un inhibidor del CYP3A4 (véase Propiedades, Farmacocinética). El cilostazol, sin embargo, no parece aumentar los niveles plasmáticos de drogas metabolizadas por el CYP3A4, ya que no tuvo efecto sobre el lovastatin, una droga cuya metabolización es muy sensible a la inhibición del CYP3A4. Riesgo cardiovascular: la administración oral repetida de cilostazol a perros (≥ 30 mg/kg/día durante 52 semanas, ≥ 150 mg/kg/día durante 13 semanas y 450 mg/kg/día durante 2 semanas) se asoció con lesiones cardiovasculares que incluyeron hemorragia endocárdica, depósitos de hemosiderina y fibrosis del ventrículo izquierdo, hemorragia en la pared de la aurícula derecha, hemorragia y necrosis del músculo liso en la pared de las arterias coronarias, engrosamiento de la íntima de las arterias coronarias y arteritis y periarteritis coronaria. No obstante, a la dosis más baja asociada con lesiones cardiovasculares en el estudio de 52 semanas, la exposición sistémica (AUC) al cilostazol libre fue menor que la observada en humanos a la dosis máxima recomendada de 100 mg 2 veces al día. Se han informado lesiones similares en perros luego de la administración de otros agentes inotrópicos positivos (incluso inhibidores de la PDE III) y/o drogas vasodilatadoras. No se observaron lesiones cardiovasculares en ratas luego de 5 a 13 semanas de administración de cilostazol en dosis de hasta 1.500 mg/kg/día. Con esta dosis, la exposición sistémica (AUC) al cilostazol libre fue de 1.5 a 5 veces (ratas macho y hembra, respectivamente) la exposición humana a la dosis máxima recomendada. Tampoco se observaron lesiones cardiovasculares en ratas luego de 52 semanas de administración de cilostazol en dosis de hasta 150 mg/kg/día (exposición sistémica al cilostazol libre alrededor de 0.5 y 5 veces en ratas macho y hembra, respectivamente, la exposición humana a la dosis máxima recomendada). Tampoco se observaron lesiones cardiovasculares en monos luego de la administración oral de cilostazol durante 13 semanas en dosis de hasta 1.800 mg/kg/día.
Sobredosificación: La información relativa a sobredosificación con cilostazol en humanos es limitada. Se puede anticipar que los signos y síntomas de sobredosificación aguda corresponderían a un excesivo efecto farmacológico: cefalea severa, diarrea, hipotensión, taquicardia y, posiblemente, arritmias cardíacas. El paciente debe ser observado cuidadosamente y se le debe proporcionar tratamiento de soporte. Dado que el cilostazol tiene una elevada unión a proteínas, es improbable que pueda ser removido en forma eficiente por hemodiálisis o diálisis peritoneal. La LD50 del cilostazol en ratones y ratas es superior a 5.0 g/kg y en perros es superior a 2.0 g/kg. Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los centros de toxicología: Hospital General de Nińos "Dr. Ricardo Gutiérrez", Tel: (011) 4962-6666 / 2247. Hospital General de Nińos "Dr. Pedro de Elizalde", Tel: (011) 4300-2115 / 4362-6063. Hospital Nacional "Prof. A. Posadas", Tel: (011) 4654-6648 / 4658-7777. Hospital de Pediatría "Sor María Ludovica", Tel: (0221) 451-5555.
Conservación: Condiciones de conservación y almacenamiento: Conservar en su envase original, a temperaturas inferiores a los 30 °C.
Observaciones: Mantenga este medicamento fuera del alcance de los nińos. Ante cualquier duda consultar al 0-800-444-2382 (BETA). Fecha de la última revisión: 04.01.
 Laboratorio
Laboratorio